
Abstract
In complex multicellular systems, gene expression is regulated at multiple stages through interconnected complex molecular pathways and regulatory networks. Transcription is the first step in gene expression and is subject to multiple layers of regulation in which epigenetic mechanisms such as DNA methylation, histone tail modifications, and chromosomal conformation play an essential role. In recent years, CRISPR-Cas9 systems have been employed to unearth this complexity and provide new insights on the contribution of chromatin dysregulation in the development of genetic diseases, as well as new tools to prevent or reverse this dysregulation. In this review, we outline the recent development of a variety of CRISPR-based epigenetic editors for targeted DNA methylation/demethylation, histone modification, and three-dimensional DNA conformational change, highlighting their relative performance and impact on gene regulation. Finally, we provide insights on the future developments aimed to accelerate our understanding of the causal relationship between epigenetic marks, genome organization, and gene regulation.
Introduction
Control of gene expression plays a key role in a wide variety of biological processes. The precise, temporal order of gene expression during development is, for instance, critical to ensure proper lineage commitment, cell fate determination, and organogenesis.1,2 However, several other processes such as cellular stress responses, tissue homeostasis, and immunity also rely on the regulation of gene expression to produce precise amounts of proteins or non-coding RNA (ncRNA) at exactly the right time and in the appropriate cell/tissue. Reflective of its importance to cell survival and function, the regulatory mechanisms controlling eukaryotic gene expression are extremely complex. 3 The total amount and the activity level of a specific protein in a cell is thus the result of the combinatorial action of transcriptional, post-transcriptional, translational, and post-translational regulatory processes. Nevertheless, the first step in this chain of events, the transcription of a gene, can be considered the most critical one. The regulatory mechanisms controlling transcription are themselves exquisitely sophisticated and rely on a complex interplay between transcription factors, chromatin-associated proteins, dynamic epigenetic modifications, and a spatially organized genome.4–7 These various components orchestrate transcriptional regulation in a complex, multilayered, and interconnected fashion.
Epigenetic processes regulate gene expression by modulating the frequency, rate, or extent of gene expression. The epigenetic machinery is composed principally of four interconnected components that ultimately define active or inactive states of chromatin: DNA methylation, histone post-translational modifications, chromatin three-dimensional (3D) conformation, and ncRNAs. Epigenetic modifications play a key role in gene expression, integrating information encoded in the genome with other molecular and chemical cues to ensure a dynamic and flexible response to intracellular and extracellular stimuli, including those from the environment. 8
In keeping with its importance, alterations of the epigenome due to the dysregulation of signaling pathways linking an environmental or physiological change with chromatin alterations and mutations of epigenetic regulators contributes to the pathogenesis of various inherited or somatically acquired human diseases, including neurological disorders, 9 autoimmune disorders, 10 diabetes, 11 and imprinting disorders. 12 In particular, many studies have shown that epigenetic changes are closely associated with each of the steps involved in cancer development and progression, including the acquisition of additional properties essential for cancer metastasis.13,14
Unlike most genetic defects, however, due to the dynamic nature of the epigenome, epigenetic defects are reversible. This reversibility is an important aspect of the epigenetic contribution to diseases and makes such diseases amenable to therapeutics.
In order to understand how chromatin dysregulation contributes to disease, as well as preventing or reversing this type of dysregulation, the levels or activities of epigenetic modifiers have been modulated through genetic manipulation (i.e., overexpression or knockout/knockdown) 15 or by preventing a DNA or histone modification from being deposited using small molecule inhibitors.16–18 These conventional approaches, however, lack gene specificity, and global chromatin changes are often induced, making it difficult to determine whether the observed phenotypes are due to a specific epigenetic change or the result of secondary global effects. Furthermore, without temporal control in altering the epigenetic marks, it becomes challenging to investigate the highly dynamic epigenetic regulatory events. 19
Recently, there has been a paradigm shift in the field, and scientists have begun investigating perturbation of the chromatin landscape in a locus-specific manner. The engineering of programmable enzymes with DNA-binding domains has enabled genome locus-specific targeting of epigenetic modifiers to alter specific local epigenetic modifications directly. In particular, the CRISPR-dCas9 system has become the most widely used epigenome-editing tool due to its high efficiency, versatility, specificity, and ease of use. Comprising a bacterially derived Cas9 protein with cognate targeting RNA moieties, the CRISPR system has proven to be a very versatile tool in recent years. With regard to epigenomic perturbance, a nuclease-deficient Cas9 (dCas9) is tethered to specific epigenetic-effector domains. Utilizing the RNA-based genome targeting of dCas9, this combination thus allows for genomically defined epigenetic modifications. Using this approach, dCas9 has been combined with activation/repression domains of several transcription factors, such as VP16, p65 and Rta, 20 HSF1, 21 and KRAB, 22 to control the gene-specific transcriptional activity.23–26 These domains promote the concomitant recruitment of chromatin-modifying proteins, additional transcription factors, and DNA-binding proteins, causing accumulation of epigenetic markers, chromatin de-condensation/condensation, and ultimately transcriptional perturbance. In order to address the demand of a better understanding of the impact of single epigenetic modifications on gene expression, as well as to help understanding the complex chromatin layer involved in transcriptional regulation, it is, however, important to design systems to fine-tune individual epigenetic perturbations. Such systems may not maximize up- or downregulation of the target gene, but they offer the advantage of precision and can potentially be employed for correction of specific disease-associated epigenetic aberrations.
Here, we review these CRISPR-based epigenetic editors for targeted DNA methylation/demethylation, histone modification, and 3D DNA conformational change.
DNA Methylation/Demethylation
DNA methylation/demethylation processes
DNA methylation is one of the major mechanisms of epigenetic regulation, playing a crucial role in many regulatory processes, including transcriptional gene expression, genomic imprinting, X inactivation, silencing of mobile elements, and maintenance of genome integrity.27–30 As a consequence, aberrant methylation plays a role in the pathogenesis of many diseases, including cancers. 31
In mammals, DNA methylation occurs almost exclusively within the context of cytosine-guanine dinucleotide (CpG)-rich regions, called CpG islands, via covalent addition of a methyl group to the fifth carbon position of cytosine residues to generate a 5-methylcytosine (5mC). The CpG islands are commonly found within promoter regions or distal regulatory elements where their methylation status contributes to gene transcriptional regulation. Cytosine DNA methylation prevents the binding of transcription factors and serves as a signal for the recruitment of epigenetic modifiers or chromatin remodeling factors, often resulting in gene silencing, though there are some notable exceptions.32,33
The 5mC mark is deposited by DNA methyltransferases (DNMTs) in a post-replication reaction with the transfer of a methyl group from S-adenosyl-
Removal of the methyl group from methylated cytosine (5mC), demethylation, can occur through either passive or active pathways. 37 Passive demethylation is replication dependent and occurs during DNA replication when maintenance methylation of a new DNA strand is inhibited, leading to concomitant loss of 5mC residues. 38 Active demethylation is a replication-independent process and involves the conversion of methyl-cytosine to cytosine via either enzymatic oxidation or deamination followed by base excision repair. The DNA methylation enzymes are dioxygenases belonging to the ten-eleven translocation (TET) family (TET1, TET2, and TET3). TET enzymes catalyze the oxidation of methyl groups and produce intermediates that are then recognized and excised by thymine DNA glycosylase (TDG). 39
Despite its well-established influence on gene expression, demonstrating an unambiguous causal relationship between DNA methylation and transcription has proven difficult. Fortunately, the recent development of gene-specific targeting tools for manipulating DNA methylation offers the opportunity to address these gaps in our understanding.
CRISPR-mediated DNA methylation
The first attempt to generate a CRISPR-based tool capable of modulating DNA methylation at a specific locus was made by Vojta et al., fusing dCas9 to the catalytic domain of DNMT3A through a flexible Gly4Ser linker 40 (Fig. 1A). Following transient transfection of HEK293 cells, the dCas9–DNMT3A fusion protein induced an increase in CpG methylation in a ∼35 bp wide region centered 27 bp downstream from the protospacer adjacent motif (PAM) sequence. Targeting a promoter with multiple gRNAs enabled a wider methylation pattern, inducing transcriptional repression of the target gene. The highest methylation activity was reached between 5 and 15 days after transfection with a dCas9–DNMT3A expressing plasmid. However, the CpG methylation achieved was not stable and was gradually lost. Following this first report, other attempts have been made to optimize the system for higher efficiency and specificity. On the basis of previous work showing that DNM3L increases the methylation activity of DNMT3A and DNMT3B, 41 Stepper et al. engineered a DNMT3A–DNMT3L chimeric protein fused to dCas9 and showed widespread DNA methylation of CpG islands in the targeted promoter at four to five times higher levels of targeted methylation than those achieved using dCas9-DNMT3a 42 (Fig. 1B). CpG methylation was observed ∼25 bp upstream and ∼40 bp downstream of the PAM site, spreading up to 1,200 bp, whereas the 20–30 bp comprising the dCas9-binding site were protected against methylation. Targeting the promoter with multiple gRNAs did not increase the methylation level. It was hypothesized that the higher methylation activity was due to multimerization of DNMT3A–DNMT3L complexes on the DNA.43,44 They also reported that DNA methylation of the targeted promoters was associated with transcriptional silencing, with only mild off-target effects. They did not report, however, whether the methylation and/or the transcriptional repression was maintained upon loss of dCas9–DNMT3A–DNMT3L. The transient nature of the transfection-based methylation gain and consequent transcriptional repression was investigated by Amabile et al. 45 They compared dCas9–KRAB and dCas9–DNMT3A fusions, showing that both systems were capable of transcriptional repression of target genes, although with different silencing kinetics. dCas9–KRAB induced rapid and robust silencing, but its effect was rapidly reversed upon loss of the dCas9–KRAB. In contrast, silencing induced by dCas9–DNMT3A was much slower and weaker than dCas9–KRAB but more stable over time, even after loss of the fusion protein. The transient co-expression of dCas9–KRAB and dCas9–DNMT3A induced rapid and long-term repression of the target genes that was further improved by co-transfection with dCas9–DNMT3L (Fig. 1C). They reconcile these observations by proposing a model whereby KRAB–KAP-1 complexes establish a chromatin environment conducive to de novo DNA methylation imposed by DNMT3A–DNMT3AL, resulting in compaction of chromatin and long-term memory of gene silencing. Finally the authors confirmed that this silencing technology is highly specific at the genome-wide level. Potential off-target DNA methylation delivered by these epigenome editing tools is indeed a concern, and widespread off-target activity of dCas9-methyltransferases, or even the catalytic domain of DNMT3a alone, has been observed. 46 The transient delivery of the epigenetic modifiers limits the residence time of these proteins, also decreasing the likelihood of off-target activity. This strategy was recently used to induce long-term methylation of insulator sequences and alterations of CTCF-mediated gene loops.47,48
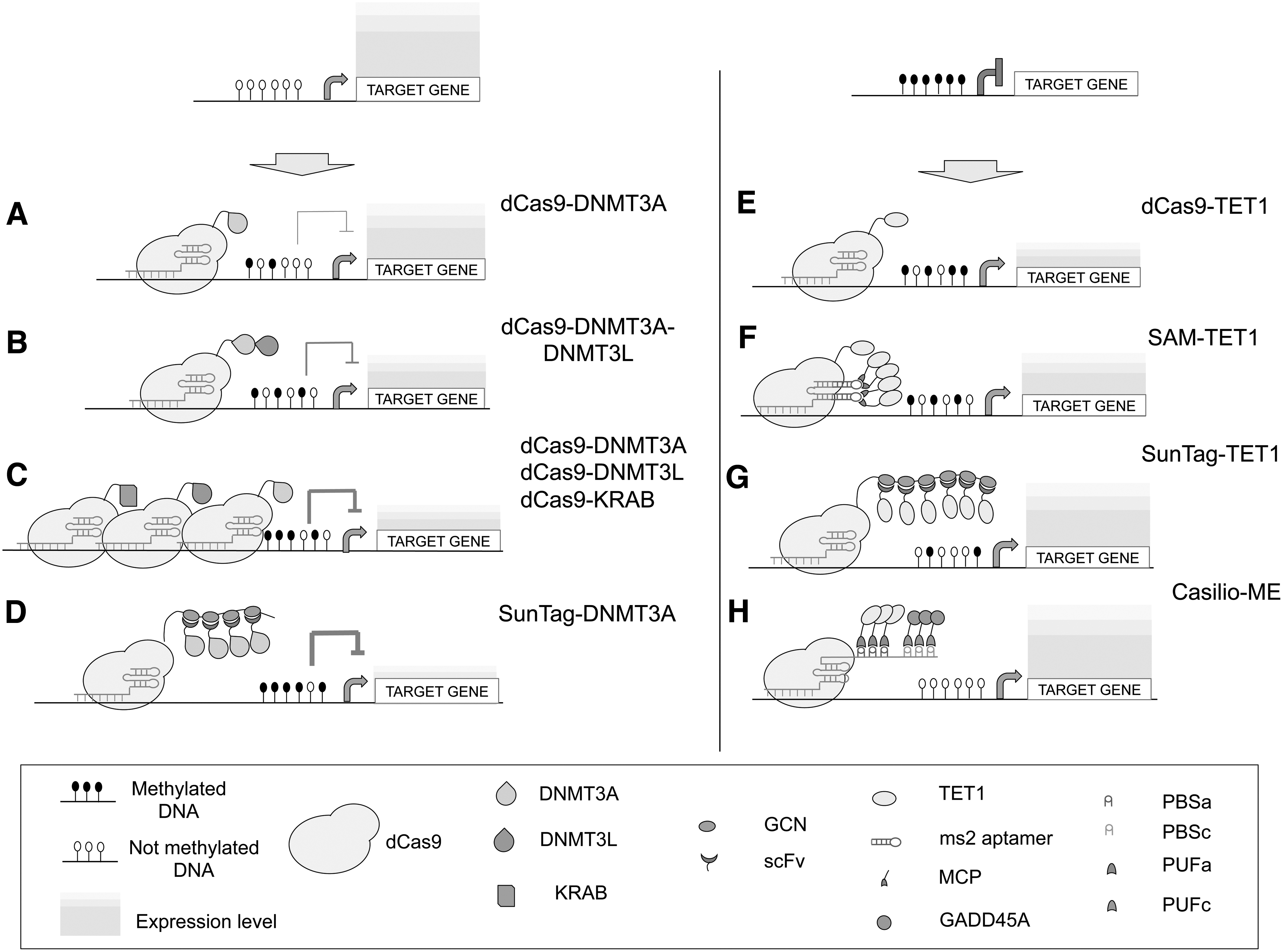
Amplification of the intended epigenetic effect can be achieved by fusing dCas9 to multiple copies of an effector domain, or targeting the DNA regions with multiple gRNAs. A third approach exemplifying this, based on the so-called SunTag system, employs a repeating peptide array with the capacity to recruit multiple copies of an scFv-fusion effector domain at a target locus. 49 Huang et al. 50 developed a dCas9–SunTag–DNMT3A system that was able to recruit multiple copies of DNMT3A to the HOXA locus in HEK293 cells obtaining higher methylation efficiency compared to the dCas9–DNMT3A fusion (Fig. 1D). In an effort to improve specificity, Pflueger et al. used the SunTag–DNMT3A system described above and modulated the expression of both the DNMT3A catalytic domain and the dCas9–SunTag. 51 In doing so, they reduced off-target activity by preventing excess of DNMT3A in the cell and reducing the availability of dCas9 to bind to off-target loci. The titration of the effector domain, however, led to a reduction of on-target activity, and target methylation might fluctuate, depending on the cell line and the expression levels of the effector domain. 51 In a recent report, Hofacker et al. tackled this problem by generating DNMT3A–DNMT3L variants containing mutations in the DNMT3A domain, which reduced the endogenous DNA binding affinity of the DNMT3A subunit. 52 The resulting SunTag–DNMT3A–DNMTL system shows highly active targeted methylation with strongly improved specificity, without the need to titrate the system components.
CRISPR-mediated DNA demethylation
Direct fusion of the TET1 catalytic domain to dCas9 represents the first tool employed for targeted demethylation of 5mC marks using the CRISPR system.48,53 In this study, Choudhury et al. used dCas9–TET1 to target the BRCA1 promoter and re-activate gene expression selectively (Fig. 1E). In other reports, the fusion protein was used to target other elements outside of gene promoters. For example, it was applied to demethylate a distal enhancer (MyoD), promoting myogenic reprogramming in fibroblasts,48,53 or to demethylate CGG repeats in fragile X syndrome-induced pluripotent stem cells, reactivating the silenced FMR1. 54
In order to improve efficiency, Xu et al. adapted a two-part RNA aptamer-derived system 21 to recruit multiple copies of TET1. 55 The system combines dCas9–TET1 with a modified single-guide RNA (sgRNA) targeting moiety containing two MS2 hairpins (MS2-sgRNA). Each MS2 hairpin interacts with one MS2 bacteriophage coat protein (MCP) fused to TET1 (Fig. 1F). This aptamer-based system showed a significant increase in demethylation and upregulation of target genes compared to dCas9–TET1 alone.
An alternative strategy to recruit multiple copies of TET1 and reach higher demethylation levels is based on the aforementioned SunTag multimerization system. Morita et al. optimized the SunTag linker from 5 to 22 amino acids, allowing more efficient recruitment of multiple copies of scFv-fused TET156 (Fig. 1H). Specificity of the SunTag–TET1 system was investigated by whole-genome bisulfite sequencing and RNA sequencing analyses indicating a low off-target activity. 56 However, careful consideration should be given to the specificity issue, as overexpression of TET1 catalytic domain alone is enough to induce demethylation and upregulation of endogenous genes. 57 Therefore, off-targets could potentially be further minimized by titrating the expression level of the scFv–TET1 fusion.
Despite the high demethylation levels reached with the SunTag system, the level and kinetic of upregulation of endogenous genes targeted with locus-specific tethering of TET1 discussed above are usually lower than those obtained fusing dCas9 with the activation domains of transcription factors. However, demethylation can have an indirect effect on gene expression such as making the target gene responsive to activation upon stimulatory events. 58 Co-recruitment of TET1 and transcription factor domains can result in a synergistic effect and higher and persistent target gene upregulation, indicating that that demethylation can create a chromatin state more permissive for programmable transcription.59–61
The employment of exogenous TET1 enzyme can cause off-target activity and/or result in other potential issues such as generation of oxidized 5-meC derivatives that may become stable independent epigenetic marks on their own and therefore perform specific unwanted regulatory functions.62,63 To tackle these problems, alternative demethylation strategies not based on TET activity have been also developed. Lu et al. used a CRISPR-dCas9 system with a modified gRNA capable of recruiting and sequestering endogenous DNMT1, thus inhibiting DNMT1 enzyme activity at the specific target site and preventing DNA methylation maintenance during replication. 64 Another potential demethylation strategy is the use of DNA glycosylase enzymes instead of TET enzymes. Gregory et al. showed that targeted DNA demethylation using TDG can indeed upregulate gene expression. 65 In a recent paper, the Casilio system was employed to achieve co-recruitment of multiple copies of TET1, along with different components of the base-excision repair machinery to the targeted methylated promoter. 66 This RNA aptamer-derived system utilized modified gRNAs containing PUF-binding sites (PBS) and PBS binding proteins fused to effector domains 67 (Fig. 1H). TDG, NEIL1, and NEIL2 are DNA glycosylases involved in the initial step of removing oxidized cytosines produced by TET1. Because initiating repair of oxidized cytosines by the base-excision repair machinery might be a limiting step to active demethylation, the Casilio system was used to couple TET1 activities with DNA glycosylases in an attempt to facilitate DNA demethylation. Surprisingly, only co-recruitment of NEIL2 increased demethylation efficiency and the activation of the target genes. On the other hand, the co-recruitment of TET1 with another factor, GADD45A, a protein involved in genome stability and DNA repair, resulted in an enhanced turnover of the oxidized cytosine intermediates and increased DNA demethylation. The authors compared the Casilio system with other platforms available for targeted DNA demethylation, including the SunTag system mentioned above, and showed increased activation of a methylation-silenced gene, presenting the Casilio system as one of the best-performing systems for targeted DNA demethylation available so far.
Finally, a plant-derived DNA glycosylase (ROS1) was recently used to remove 5-meC directly without prior modifications, avoiding formation of 5-meC derivatives. 68
Histone Modification
Histone post-translational modifications and gene expression
A major mechanism by which chromatin structure and function are regulated is through the actions of reversible, post-translational histone chemical alterations. An astonishing number of modifications, including acetylation, methylation, citrullination, ubiquitination, SUMOylation, ADP-ribosylation, proline isomerization, and phosphorylation, occur on histones. 69 While the majority are found in the flexible N- and C-terminal “tail” domains that protrude away from the nucleosome core particle, a significant number also occur in the histone globular domains that regulate histone–histone and histone–DNA interactions. The pattern of modifications has been suggested to act as an information code, the so-called histone code.70,71 Distinct histone marks function sequentially or in combination to initiate distinct downstream changes dictating both nucleosomal interactions and the association of non-histone proteins that collectively influence packaging and gene regulation. Furthermore, histone modifications can interact with DNA methylation and together give rise to an epigenetic code associated with high or low levels of gene expression. 72
Histone modification are imposed, removed, or recognized by specific proteins. “Writers” are enzymes that deposit the histone modification, “erasers” are enzymes that remove the modification, and “readers” are those protein domains present in effector proteins, such as transcription factors, which recognize a modification and initiate downstream processes.
The most studied histone modifications are methylation and acetylation of lysine residues in the N-terminal tails of histones H3 and H4, as they appear to play a major role in dictating the chromatin state and thus the transcriptional state. 69 The lysine residues on histone tails can be mono-, di-, or tri-methylated by a set of histone writer proteins, generically referred to as histone methyltransferases (HMT) and removed by histone demethylase (HDM). The transcriptional influence of lysine methylation is strongly context dependent. However, methylation of lysine 4 on histone 3 (H3K4) and lysine 36 (H3K36) are often associated with gene activation.73–75 In contrast, modifications, such as methylation of lysine 9 on histone 3 (H3K9), lysine 27 (H3K27), or lysine 20 (H3K20), are often linked to silenced genes.73,76,77 Lysine residues such as lysine 36 (H3K36) and lysine 64 (H3K64) on histone 3 can be acetylated, which typically results in a less condensed chromatin state coupled with an increase in transcriptional activity.78–80 Histone acetyl transferases (HATs) are the writer enzymes that deposit acetylation marks, and histone deacetylases (HDACs) are the eraser enzymes.
Open questions regarding correlation versus causation between epigenetic modifications and gene regulation still remain unanswered. Are the actual histone marks required for changes in transcription, or are those modifications only passengers? Can specific combinations of modifications be undoubtedly associated to specific outcomes opening the road toward the deciphering of the histone code?
For CRISPR-based epigenetic editors, the catalytic domains of HATs, HDACs, HMTs, and HDMs can be fused or non-covalently recruited to dCas9. These synthetic editors represent a new tool for the programmed manipulation of the epigenetic landscape and offer the opportunity of studying interactions between different epigenetic marks at specific loci.
CRISPR-mediated histone methylation
Targeted histone methylation can be achieved fusing dCas9 with proteins such as KRAB or FOG1, which in turn recruit nucleosome modifying complexes resulting in acquisition of H3K9me322 and H3K27me3, 81 respectively, and loss of histone acetylation.
However, a more direct approach for targeted histone methylation consists of a direct fusion of the catalytic domain of specific HMTs to dCas9 (Fig. 2). Fusing dCas9 to the catalytic domains of HMTs PRDM9 and DOT1L, Cano-Rodriguez et al. show how the interplay between different histone marks and the preexisting chromatin microenvironment can impact the stability of acquired epigenetic marks and the transcriptional status of the target gene. In their study, the H3K79me mark, introduced by dCas9–DOT1L, seems to be required for the stability and maintenance of the H3K4me3 modification introduced by dCas9–PRDM9, and the long-term effects this editing has on gene activation depends on the methylation level of the targeted DNA. 82
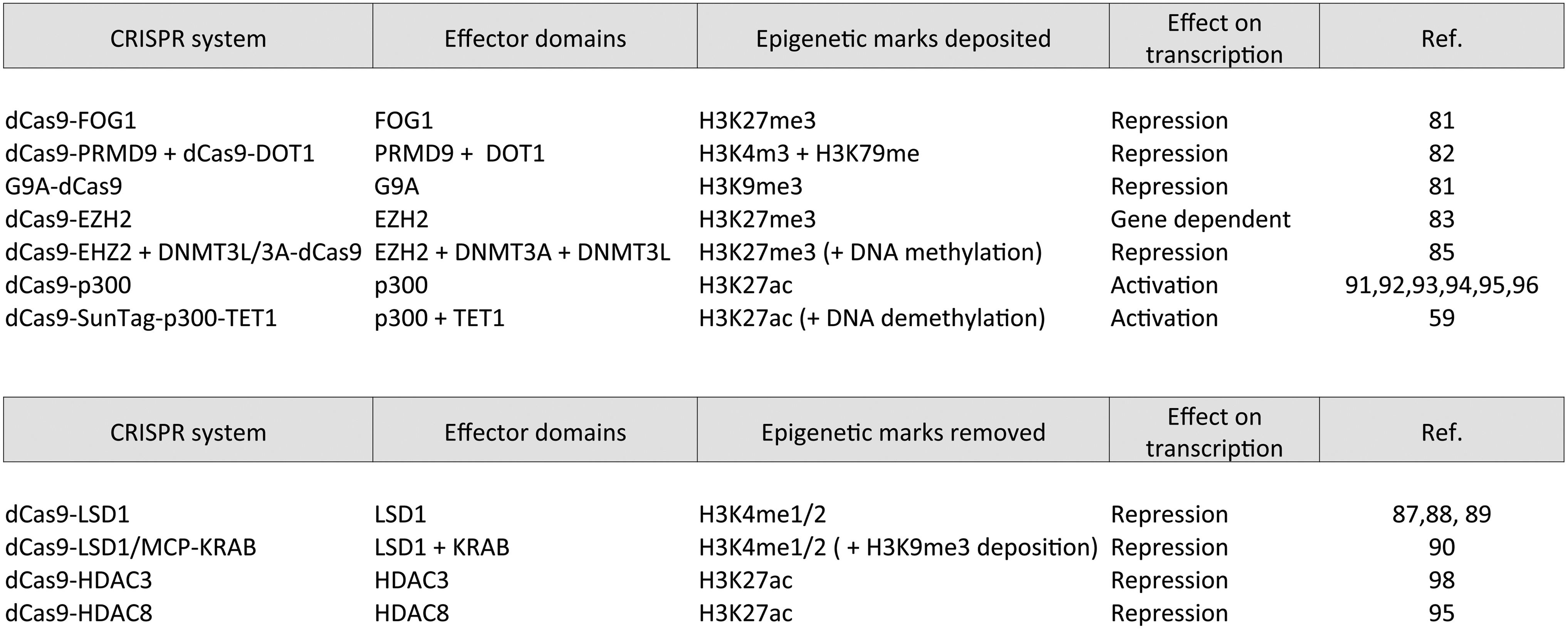
CRISPR systems employed for targeted histone modifications.
Other HMTs were fused to dCas9 to investigate the causal relationship between the presence of a specific histone mark and altered gene expression. 81 Downregulation of HER2 was observed following the recruitment of dCas9 fused with the catalytic domains of G9A or SUV39H1 to its promoter. 81 However, whilst G9A–dCas9 deposited the expected repressive H3K9me3 mark, the recruitment of SUV39H1–dCas9 did not result in H3K9me3 addition or alternative histone modifications, implicating steric hindrance of SUV39H1–dCas9 as responsible for target gene repression. A similar effect was observed when fusing dCas9 to the full-length methyltransferase EZH2. These results highlight the importance of using catalytic dead epigenetic effectors as controls to assess the impact of epigenetic marks on gene expression and exclude the steric hindrance of dCas9 fusion proteins as cause of transcriptional interference.
EZH2 is the catalytic subunit of the Polycomb Repressive Complex 2, and it is responsible of deposition of H3K27me3, an epigenetic mark often associated with gene silencing. The effector domain EZH2 was tested in two other reports. dCas9–EZH2 deposited H3K27me3 at specific targeted loci, inducing gene repression, but the effect of H3K27me3 on transcription was gene dependent. 83 Interesting, the authors show that when the DNA target is methylated, dCas9–EZH2 can still bind to the target region but is unable to methylate H3K27, suggesting an antagonism between DNA methylation and H3K27 methylation. 84 In contrast, a synergism between DNA methylation and H3K27me3 was reported in another recent paper. 85 In this work, the combination of induced DNA methylation and histone methylation obtained by co-recruitment of DNMT3L–DNMT3A–dCas9 and EZH2–dCas9, resulted in the long-term repression of the target gene HER2. However, the effect was gene dependent: long-term repression of alternative target genes not obtained by combining DNMTs and EZH2 was instead achieved by co-recruitment of DNMTs and KRAB (that itself recruits a complex containing a histone methylase), as previously reported. 45 The authors conclude that long-term epigenetic memory can be achieved, but different histone and DNA methyltransferases might be required to obtain long-term repression at different loci and/or in different cell types. All these data once more show the complex cooperation between different epigenetic marks and the preexisting chromatin context in modulating gene expression.
CRISPR-mediated histone demethylation
LSD1 is a HDM that catalyzes the removal of the methyl groups from H3K4me1/2 and H3K9me2, which are associated with active and repressive chromatin, respectively. 86 LSD1 was used as a TALE-fused effector to demethylate enhancer-associated histone methylation, leading to inactivation of targeted enhancers. 87 Subsequently, LSD1 or KRAB were coupled to Neisseria meningitidis dCas9 and used in mouse embryonic stem cells to target known enhancers of Oct4 and Tbx2 and their promoters. 88 Both epigenetic modifiers had a negative impact on gene expression but with different modalities of action. dCas9–LSD1 efficiently interfered with the initiation of the enhancer activity. However, when targeted to the promoter region, it did not cause any change in the histone landscape and had no effect on gene expression. On the contrary, dCas9–KRAB was capable of repressing already engaged regulatory elements and silencing promoter activity. These observations were confirmed in another report, 89 supporting the hypothesis that dCas9–LSD1 can efficiently interfere with enhancer activity only at early steps of gene activation, whereas dCas9–KRAB has a more potent repression activity at genes already engaged in transcription. Recently, the combinatorial action of LSD1 and KRAB in repressing enhancer activity was tested using an aptamer-based strategy combining dCas9–LSD1 together with MS2-sgRNA to recruit MCP–KRAB repressor domains 90 (Fig. 2). Targeting the β-globin enhancer HS2 in K562 erythroleukemia cells, the authors showed a stronger repression of β-globin genes using dCas9–LSD1/MCP–KRAB compared to the single-effectors dCas9–KRAB or dCas9–LSD1. dCas9–LSD1/MCP–KRAB led to concurrent and more significant increase in H3K9me3 and decrease in H3K4me1/2 compared to dCas9–KRAB or dCas9–LSD1 alone. Furthermore, enhancer repression caused epigenetic changes at both targeted enhancers and associated gene promoters likely through enhancer–promoter interactions. The dCas9–LSD1/MCP–KRAB combination was used in vivo to determine the functional role of lineage-specific enhancers required for lineage differentiation of HSCs in mouse. 90
CRISPR-mediated histone acetylation
The most commonly used HAT family in the field of epigenetic biotechnology is the p300/CBP family of HATs. The p300 catalytic domain fused to dCas9 enables acetylation of H3K27 in a locus-specific manner. Recruitment of dCas9–p300 to either promoters or enhancers induces target gene activation concomitant with the deposition of H3K27ac marker. 91 As alternative to dCas9, a dCpf1–p300 fusion protein was also successfully used to activate transcription from either promoters or enhancers in human cells. 92 dCas9–p300 has been employed to screen for gain of regulatory element function that is otherwise difficult to obtain with genome editing. 93 These high-throughput screens facilitate the identification of both proximal and distal transcriptional regulatory elements in their putative chromosomal context and could potentially be used to screen for regulatory elements that affect cellular phenotype, including drug resistance or cell growth. dCas9–p300 can also work as a transcriptional reporter to screen small molecules for their ability to disrupt the HAT-dependent gene expression. 94 A recent paper provides another excellent example of applications of epigenetic modifiers; dCas9–p300 and dCas9–HDAC8 were used in primary neurons to target the enhancers of Fos, a transcription factor responsible for coupling short-term extracellular signals to long-term changes in neuronal phenotype. 95 Using these epigenetic tools, the authors show a correlation between the histone acetylation level of the targeted enhancer and the transcriptional kinetics of the Fos gene, with higher acetylation levels resulting in an increased transcriptional burst frequency in response to membrane depolarization. This paper provides a clear demonstration of how chromatin regulators can fine-tune the dynamic features of stimulus-inducible gene transcription. dCas9–p300 has also been used in primary T-cell to activate and stabilize the expression of Foxp3. 96 Recently, the additional effect of p300 and VP64 co-recruitment in activating targeted enhancers was obtained by combining dCas9–p300 together with MCP–VP64 in an aptamer-based recruitment strategy. 90 Synergistic effect between histone acetylation and DNA demethylation was also obtained by combining p300 with TET1 catalytic domains using a SunTag system, although the effects were gene dependent 59 (Fig. 2). These few reports infer that the combinatorial action of different epigenetic modifiers in a locus-specific manner can be easily achieved using the CRISPR-epigenetic systems, providing a framework for studying the effect of multiple epigenetic markers on gene regulation.
CRISPR-mediated histone deacetylation
HDACs are the eraser enzymes that remove lysine acetylation, often resulting in transcriptional repression. The enzymes belonging to the class I HDAC family (HDAC1/2/3/8) are ubiquitously expressed and form part of repressive protein complexes, and for these reasons are often employed in the field of epigenetic biotechnology. 97 Full-length human HDAC3 was fused to dCas9 and used to target the promoter of target endogenous genes. 98 dCas9–HDAC3 was able to repress gene transcription following deacetylation of H3K27ac. However, the effect on transcription was relatively modest and ineffective at regions with high enrichment of H3K27ac. dCas9 fused to HDAC8 was successfully used in primary neurons to target an enhancer element of the Fos gene. 95 Recruitment of dCas9–HDAC8 reduced H3K27ac locally and significantly decreased Fos expression both under basal conditions and following membrane depolarization. Furthermore, enhancer histone deacetylation decreased the overall frequency of Fos transcriptional bursts upon stimulation, suggesting a functional role of chromatin modifications in fine-tuning the transcriptional level of stimulus-inducible genes.
Programmable 3D Genome Architecture Modifications
3D genome architecture and gene regulation
Regulation of gene expression in eukaryotes is extremely complex as a result of different regulatory layers. 3 Epigenetic processes such as chromatin remodeling and DNA methylation are intimately linked to the 3D genome architecture. 99 Chromosome location and structure have implications for gene regulation, and the dynamic reorganization of genome topology seems to have an instructive role in implementing transcriptional programs in response to environmental clues.100,101 There are different levels of chromatin organization. Individual chromosomes occupy distinct territories that form interchromosomal hubs, and within chromosome territories, active and inactive chromatin segregate into megabase-scale A and B compartments. 102 The A compartments are highly accessible regions marked by histone modifications associated with regulatory activity and are typically associated with a more central nuclear position. B compartments are regions of low accessibility marked by repressive histone marks and are enriched at the nuclear periphery/lamina. 103 On a sub-megabase level, chromatin is organized into insulated spatial domains named topologically associating domains (TADs). 104 Within these domains, gene expression is controlled by regulatory elements involving dynamic interactions between promoters and enhancers that are relatively restrained by the boundaries of the TAD or loop domain in which they reside.105,106 To be activated by an enhancer, a gene should therefore be located within the same TAD in which the enhancer resides. Enhancers are usually located far away from the target gene, and their interaction occurs through the looping of the intervening DNA segment. A gene can be regulated by the combined action of several enhancers located within the same TAD, increasing the regulatory capacity of eukaryotic cells. Several DNA-binding proteins such as CTCF, cohesion, and transcription factors are involved in determining the boundaries of these domains and in generating chromatin loops.107,108
CRISPR-mediated programmable 3D genome architecture modifications
Several groups have assessed manipulation of nuclear architecture using CRISPR-based systems. CRISPR-mediated disruption of CTCF-binding sites is one way to create topological changes 109 (Fig. 3C). However, more fine-tuned, reversible, and less disruptive ways to re-engineer genome conformation have recently been proposed. Wang et al. designed a CRISPR-based system to achieve chemically inducible recruitment of endogenous genomic loci to distinct nuclear compartments. 110 This system, called CRISPR-genome organizer (CRISPR-GO), offers an inducible and reversible approach to study spatial genome organization and its impact on gene regulation (Fig. 3A). A similar system was recently tested in yeast, exploiting the interaction between cohesin and dockerin. By fusing cohesin to dCas9 and dockerin to the nuclear membrane protein Esc1p, they were able to localize desired genes to the nuclear periphery. 111 The CLOuD9 system developed by Morgan et al. is a CRISPR-based system to provoke chemically inducible targeted chromatin loop formation, providing an important tool for studying the impact of long-range interactions between enhancers and promoters on gene expression 112 (Fig. 3B). They demonstrate that chromatin looping alone is sufficient to alter gene expression. The chromatin loop reorganization is reversible after ligand removal, but stable chromatin loops could be achieved after prolonged ligand exposure. Using CLOuD9, the authors identified a novel mechanism for stable formation of de novo chromatin contacts independent of cohesin and CTCF. These examples clearly show how CRISPR-based systems can be used to facilitate a deeper understanding on the role of large-scale chromatin organization in the control of transcriptional dynamics. It is also becoming increasingly clear that 3D genome alterations can have pathological consequences.113,114 The disruption of TAD boundaries usually affects transcription regulation and may be the cause of various diseases, including cancer.115,116 Cancer cells usually acquire new TAD boundaries or lose preexisting ones, depending of the type of cancer.117,118 These alterations are often followed by changes in the epigenome and formation of new cancer-specific chromatin loops, leading to transcriptional activation of oncogene and/or repression of tumor suppressors. The direct contribution of this mechanism to cancer development was demonstrated in vitro using CRISPR-Cas9 technique.117,119 Several efforts have aimed to develop 3D genome-modulating agents as anticancer epigenetic drugs. 113 These drugs have as target endogenous proteins that maintain the 3D genome architecture, and the lack of specificity can result in genome-wide 3D reorganization with toxic effect also on healthy cells. CRISPR-based systems are valuable tools that provide new insights into spatial genome organization and the correlation between 3D genome alterations and diseases. Furthermore, they can potentially become a new therapeutic approach for a highly targeted modulation of the genome in cancer cells.
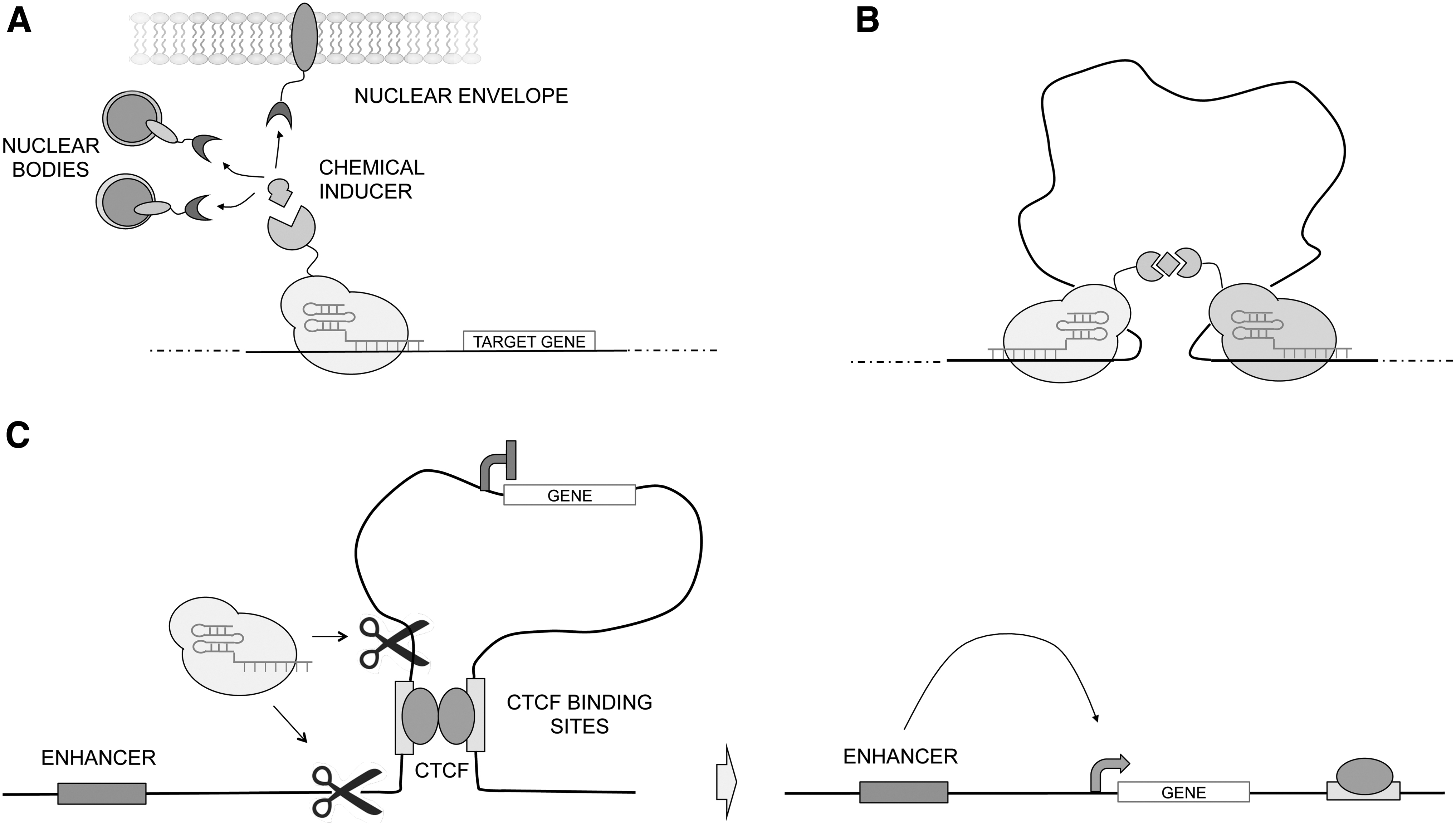
Programmable three-dimensional genome architecture modifications.
Future Directions
The CRISPR-based systems described above are powerful tools for targeted epigenetic modifications. However, they are to be employed in the sophisticated and complex arena of the eukaryotic gene regulatory landscape. Thus, several aspects must be considered for future applications: the required fine-tuning of the CRISPR modifiers, their combinatorial actions, their employment in cell type specific backgrounds, and their interactions with already existing markers or topological chromatin domains.
The fine-tuning of the CRISPR tools can be achieved by employing the new modular cloning platforms designed to generate rapidly purpose-built customized vectors, optimize new CRISPR systems, and build complex genetic circuits to gain further spatiotemporal control in different cellular types.120–123 In building new CRISPR-based epigenetic modifiers, variability in experimental results must be expected as a consequence of structural variations in the design of synthetic epigenetic modifiers. Linkers of different lengths connecting functional domains, the choice of C- or N-terminal fusion proteins, the use of alternative nucleases, or the implementation of alternative CRISPR systems should be tested in parallel to gain more insight into the impact that such variations may have on the efficiency of the epigenetic modifications. In this regard, a modular cloning system (Fig. 4) would allow for an easy and rapid generation of libraries of epigenetic modifiers variants exploring all the structural combinatorial space. 121 The expression of an endogenous gene is highly regulated and often cell-type specific. Such fine-tuned expression is the result of synergistic/antagonistic action of multiple epigenetic modifications and other complex regulatory layers. Targeting an endogenous gene with a synthetic epigenetic modifier must therefore take into account potential confounding effects derived from already existing cell-type specific epigenetic marks and secondary phenotypic effects due to variations in the expression of the target endogenous gene. Dissecting the complex regulation of endogenous genes is one way to elucidate the role of specific epigenetic marks or 3D DNA conformational changes. On the other hand, a bottom-up approach that makes use of an orthogonal and less complex environment on which to build new regulatory layers might represent a complementary approach. A good candidate for an orthogonal platform, on which studying the combinatorial effect of different epigenetic marks without messing up with endogenous genomic loci, is represented by artificial chromosomes. 122 The DNA sequence of an artificial chromosome, which persists in the cell independently from the genome, can be designed in silico to contain user-specific regulatory sequences and might therefore provide a more suitable platform for exploring the effect of epigenetic marks on the expression of gene reporters, without affecting other cellular processes and without interfering with preexisting regulatory layers. Due to their size potential, artificial chromosomes would be suitable for use as a vector for delivering both the necessary expression cassettes encoding for synthetic epigenetic modifiers and the target genes. As such, this represents a convenient surrogate methodology for empirical investigation. It could also facilitate the delivery of complex genetic circuits encoding for more sophisticated inducible CRISPR systems. Finally, it could enable investigation into long-range interactions between regulatory elements in 3D DNA conformational changes and their impact on epigenetic mark distribution (Fig. 4). Artificial chromosomes are progressively becoming easier to generate, 124 although substantial challenges in the delivery into the target cells still represent an obstacle and limit their wider use.

Synthetic biology approach to investigate the epigenome.
Finally, it is expected that a wider assortment of chromatin regulatory activities will be included in the repertoire of synthetic epigenetic modifiers, as currently only a subset of the wide array of chromatin regulatory enzymes has been tested by these approaches. For instance, the phosphorylation of histones is highly dynamic, and it takes place predominantly, but not exclusively, at N-terminal histone tails. 69 An extensive cross talk exists between phosphorylation and other posttranslational modifications, which together regulate various biological processes, including gene transcription, DNA repair, and cell cycle progression. 125 Deimination, the conversion of arginine to citrulline, effectively neutralizes the positive charge of the arginine, since citrulline is neutral, thus impacting chromatin structure.126,127 Histones can also be mono- and poly-ADP ribosylated on glutamate and arginine residues, and once again this modification is reversible. 128 Like the above-mentioned modifications, relatively little is known about the functional consequence. Histone ubiquitylation results in a much larger covalent modification but still remains highly dynamic, and it seems to have an impact on gene expression.129,130 SUMOylation has been detected on all four core histones and seems to function by antagonizing acetylation and ubiquitylation. 131 It is clear that more work is needed to elucidate the molecular mechanisms through which these and other modifications exert their effect on chromatin, and synthetic epigenetic modifiers may prove critical to empirical deciphering of the histone code.
Conclusion
In multicellular eukaryotic systems, gene expression is the result of a very complex process achieved by the coordinated action of multiple layers of regulation. Our understanding of the endogenous mechanisms that regulate gene expression is still limited. However, CRISPR-Cas9 has revolutionized gene editing and is enabling researchers to dissect the complexity of mammalian gene expression and determine causal relationships between individual epigenetic marks and gene transcription activity. The results obtained in recent years are encouraging. However, despite the progress achieved, our knowledge on chromatin regulation, maintenance, and its dynamics in a given cell type or a specific context is still in its infancy. The lack of predictive capacity and the inability to control epigenome editing reliably hinders the application of CRISPR-based epigenomic therapy for treatment of pathologies such as cancer. New CRISPR systems are offering promising solutions to unravel the complexities of epigenetic regulation further. Small molecules or light inducible CRISPR platforms provide researchers with new tools for high-resolution, temporal manipulation of epigenetic marks, enabling quantitative studies of the kinetics and dynamics of epigenetic processes. 132 In addition, orthogonal CRISPR systems can be used to edit multiple epigenetic marks simultaneously within the same cell, offering an opportunity to explore the complicated interactions between various epigenetic modifications and the 3D architecture of the genome. Integration with advances in other scientific areas such as synthetic biology will be critical to overcome challenges. 133 The future for the explorative field of epigenetic regulation seems bright. The new advancements in the field of gene editing, the result of human ingenuity, will eventually shed light in this marvelous and complex world of gene regulation.
Footnotes
Author Disclosure Statement
All authors declare no competing financial interests.
Funding Information
No funding was received for this article.