
Abstract
Recombinant adenoviruses have broad applications for gene delivery and expression. Furthermore, the adenovirus packaging system facilitates the expression of RNA-guided CRISPR/Cas9 nuclease complexes. In this study, we developed a novel system, named AdBlue, for the construction of recombinant adenoviruses using an enzymatic assembly strategy. This system could significantly reduce the time and labor required to generate adenoviral vectors. When applied to CRISPR/Cas9 design, it simplifies the preparation of recombinant adenoviruses carrying nuclease complexes and can induce high levels of site-specific mutagenesis. Our system has outstanding advantages for adenovirus preparation and could be a useful molecular engineering tool for gene delivery and editing.
Introduction
In general, four viruses, adenovirus, lentivirus, retrovirus, and adeno-associated virus, mediate the virus-based gene delivery system. Among them, adenovirus-based gene delivery system has many pros. It can infect a broad spectrum of cell types without integration into the genome of target cells, which facilitates gene transfer. Importantly, the adenovirus packaging system is feasible for generating high-titer recombinant viruses and driving high-target gene expression.1,2 However, adenoviruses-mediated gene delivery system does not integrate exogenous gene into the genome of host cells, and this limits its application in gene therapy.3,4 CRISPR/Cas9 system, a widely used gene editing tool, can modify the genome of target cells through expressing CRISPR/Cas9 nuclease complexes, whereas long-term expression of Cas9 is responsible for high rate of DNA damage. Thus, based on the duration of gene expression, adenovirus particles have great promise as agents for CRISPR/Cas9 complex gene delivery.5,6 Adenoviral vectors also work well with the novel genome-editing CRISPR/Cas9 system.7–10 Thus, genome editing using adenoviral vectors has considerable potential for gene therapy.
In tradition, two strategies have been used to generate recombinant adenoviruses.1,11–13 One is to directly insert a foreign gene into the adenoviral genome, and the second method depends on homologous recombination in mammalian cells or in Escherichia coli. The large adenovirus genome (∼36 kb) and its limited number of restriction sites restrict the application of the direct ligation strategy. However, indirect construction strategies can resolve some of those limitations. In the AdEasy system, a target gene is inserted into the Shuttle vector, then integrated with the adenovirus genome through homologous recombination in BJ5183 cells. This system overcomes the drawbacks of using the large adenovirus genome in vitro and makes it easier to generate recombination adenoviruses. 1 However, it is accompanied by difficulty screening for homologous recombination (identified by colony shape or size) and requires the construction of multiple plasmids, resulting in a time-consuming process with a high work load. Particularly, the system is inefficient for use with the CRISPR/Cas9 system, for which effective plasmids with multiple single guide RNAs (sgRNAs) must be established and screened.14,15
In 2009, Gibson et al. described an isothermal single-reaction method for assembling multiple linear DNA fragments through the concerted action of a 5′ exonuclease and a DNA ligase. This method can be used to seamlessly construct synthetic and natural genes, genetic pathways, and entire genomes without relying on specific restriction enzyme sites.16,17 To further accelerate the construction of a recombinant adenovirus genome, we modified Gibson's design to employ enzymatic assembly during target gene integration into the adenovirus genome. In this study, we demonstrate the features and advantages of our new system, which we named the AdBlue system.
Materials and Methods
Cell culture and transfection
Human embryonic kidney (AD-293) cells and rat embryonic cardiomyocytes (H9C2) purchased from ATCC were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco). The AD-293 cells constitutively expressed the AdEasy-deleted E1 gene in trans. A standard transfection with Lipofectamine™ 3000 (Thermo Fisher) was performed according to the manufacturer's instructions. Fluorescence signals were recorded by confocal microscopy (Leica SP5) after transfection.
Bacteria and plasmids
The E. coli strain TOP10 was purchased from TransGen Biotech, and transformation was performed according to the manufacturer's instructions. The adenovirus backbone vector (pAdEasy) and pShuttle plasmid were obtained from Stratagene. Extraction of plasmids and other small DNAs was performed using the Axygen® extraction kit. Large DNAs (enzyme-digested AdEasy-1 and AdBlue vectors) were extracted using phenol–chloroform extraction. The restriction endonucleases used in this study were obtained from New England Biolabs (NEB).
Polymerase chain reaction
For all polymerase chain reaction (PCR) amplifications, Q5® High-Fidelity DNA Polymerase (NEB, M0491) was used according to the manufacturer's instructions. For reverse transcription PCR, total RNA was isolated from cells with TRIzol™ reagent (Invitrogen) according to the manufacturer's protocol. RNA was reverse transcribed with the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Bio).
Enzymatic assembly
All of the enzymatic assemblies used in this study followed the protocol shown hereunder. Each reaction comprised 15 μL reaction buffer (4.8 μL Q5 buffer [NEB M0491]; 4.8 μL 25% PEG8000 [Sigma-Aldrich]; 0.6 μL T5 Exonuclease [NEB M0360]; 1.6 μL Taq DNA Ligase [NEB M0208]; 0.3 μL Q5 High-Fidelity DNA Polymerase [NEB M0491]; 2.4 μL dNTP, 2.5 mM each [NEB]; and 0.5 μL NAD [NEB B9007]) and 9 μL DNA fragments (∼10–100 ng of each DNA segment in equimolar amounts). The total 24-μL reaction was incubated at 50°C for 1 h, and then used for transformation.
Western blot analysis
H9C2 cells were seeded in six-well plates. Cells were harvested in 500 μL lysis buffer (60 mM TrisCl [pH 6.8], 10% glycerol, 2% sodium dodecyl sulfate, 5% 2-mercaptoethanol, and 0.02% bromophenol blue) 6 days after transfection. Proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene fluoride membranes (Millipore). The membranes were blocked with 5% bovine serum albumin and then incubated with primary antibodies. Subsequently, the membranes were washed three times and incubated with the monoclonal peroxidase-conjugated goat antirabbit antibody (Beyotime) diluted with blocking buffer (final dilution 1:10,000). For protein detection, we washed the blots three times, added the ECL reagent (GE Healthcare), and detected peroxidase activity according to the manufacturer's instructions. The primary antibodies used in this study were anti-CUGBP1 (Eptomics) and β-actin (Beyotime). Relative protein expression levels were analyzed by densitometry using ImageJ software (National Institutes of Health).
Production of recombinant adenoviruses
PacI-digested adenoviral plasmids were transfected into AD-293 cells and fluorescence signals were verified 2 days after transfection. Cells were cultured at 37°C, and the medium was changed every day until plaques were observed. The cells were harvested and pelleted by low-speed centrifugation, then the viral particles were liberated by three freeze–thaw cycles. To generate viral stocks with a higher titer, AD-293 cells were infected with the cell lysate and the harvest process was repeated.
sgRNA design and functional testing
We used the CRISPR design tool (http://crispr.mit.edu) to create sgRNAs. The 20-bp sequence (5′-GAAGAGTGCCGGATATTGCG-3′) in the sixth exon of the rat CUGBP Elav-like family member 1 (Celf1) gene was selected as the sgRNA target site. Seven days after infection with the sgRNA and Cas9 viruses, H9C2 cells were lysed in lysis buffer (10 mM tris [pH 8.0], 10 mM NaCl, 10 mM ethylenediaminetetraacetic acid, 0.5 mM sodium dodecyl sulfate, 1 μg/μL proteinase K, and 50 ng/μL RNase A) at 55°C for 1 h. Genomic DNA was extracted using the chloroform–phenol method. Functional testing of sgRNAs was performed with the T7E1 nuclease assay. In brief, 1000-bp genomic fragments containing the sgRNA target sites were amplified by PCR, followed by denaturation/reannealing. The products were incubated with T7 endonuclease I (NEB) at 37°C for 30 min, followed by electrophoresis on a 2% DNA gel. To confirm that the insertion–deletion mutations were located in the target site, the amplified fragments were sequenced.
Quantification and statistical analysis
Statistical analysis of the data was performed using a two-tailed Student's t test. The data are presented as the mean ± standard error of the mean.
Results
Overview of the AdBlue System
A schematic overview of recombinant adenovirus production is shown in Figure 1A. The Blue vector carrying the gene of interest (GOI) was digested with EcoRI and the AdBlue vector was digested with ClaI. Then the linearized fragments were collected through phenol–chloroform extraction. The two linearized fragments shared an ∼200-bp terminal overlap sequence (red dotted line in Fig. 1A). They were ligated together into one sealed plasmid with a one-step enzyme-catalyzed reaction. The recombinant construct was transformed into TOP10 competent cells to facilitate selection of kanamycin-resistant transformants (blue spots) for further amplification (over 90% transformation efficiency rate). We named the new sealed plasmids using the formula “AdBlue-GOI.” We digested the sealed plasmids with PacI after purification to expose the inverted terminal repeats, and then transfected the digestion products into AD-293 cells. In comparison, the AdEasy system adopts the homologous recombination machinery in E. coli to generate recombinant adenovirus genome DNA through two recombination events between the adenoviral skeleton plasmid vector and the Shuttle vector carrying the GOI (Fig. 1B).

Overview of the AdBlue System.
Construction of the AdBlue vector and the Blue vector
We inserted the lacZ gene between the right inverted terminal repeat and pBR322 origin cassettes of AdEasy-1 to generate the AdBlue vector. As the AdEasy-1 vector has a large base mass (∼30 kb) and no restriction enzyme cleavage sites, we used enzymatic assembly to insert the lacZ gene. An ampicillin resistance gene and pBR322 origin cassette were ligated into the lacZ gene carrying the ClaI restriction site (Fig. 2A) through fusion PCR. The new fusion fragment and digested AdEasy-1 vector (PacI and ClaI) shared short identical terminal sequences (red box in Fig. 2A). The integration of the two linear fragments through enzymatic assembly completed the construction of the AdBlue vector (Fig. 2A and Supplementary Data S1). The assembly products were transformed into TOP10 cells, and the colonies were subjected to blue–white screening, followed by expansion culture. Plasmids were extracted and identified by restriction analysis (ClaI digestion), and two electrophoretic bands were observed: ∼30 and ∼3–4 kb (Fig. 2C).
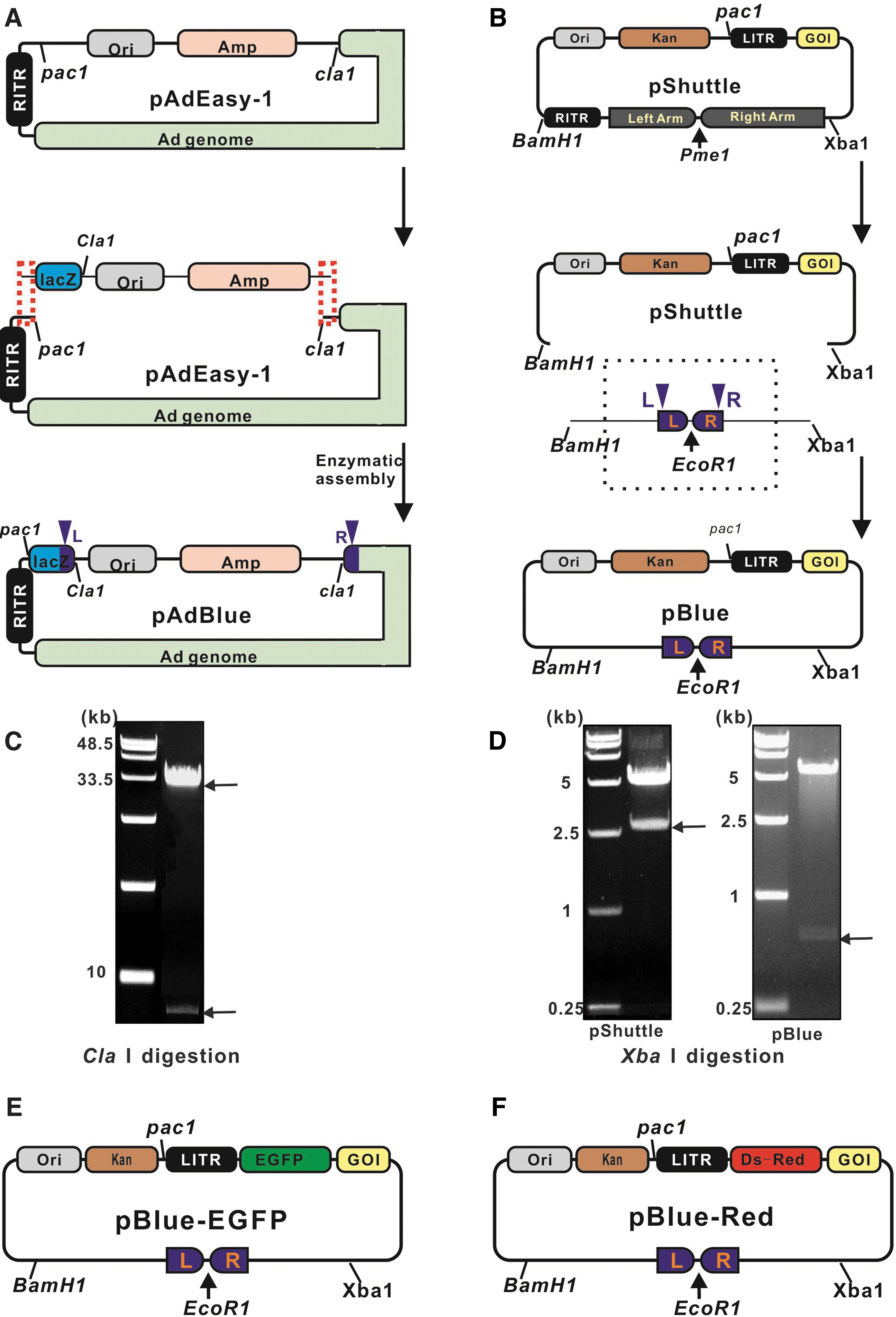
Construction of the AdBlue and Blue vectors.
To construct the Blue vector, the Shuttle vector was digested with BamHI and XbaI, and the larger fragment was extracted as the skeleton of the Blue vector. Then, the two short fragments amplified from the AdBlue vector were fused with the terminus of the lacZ gene and the linear adenoviral genome (dark blue box in Fig. 2A, marked L and R). The two newly formed fragments were subjected to fusion PCR to facilitate integration, and three restriction endonuclease cutting sites (for EcoRI, BamHI, and XbaI) were retained in the integrated fragment (Fig. 2B). Then, this newly constructed fragment was ligated into the extracted Shuttle vector backbone with T4 DNA ligase to generate the Blue vector (Fig. 2B and Supplementary Data S2), and the DNA ligation products were transformed into TOP10 competent cells accompanying with over 90% transformation efficiency rate. Transformants were selected for kanamycin resistance, and the Blue vector was subsequently identified by double restriction digestion with BamHI and XbaI. Unlike the Shuttle vector, the Blue vector was digested into a backbone of ∼5 kb and a small fragment of ∼500 bp (Fig. 2D). By inserting the enhanced green fluorescent protein (EGFP) and DsRed fluorescent protein genes into the Blue vector through multiS one step cloning kit (Vazyme), we developed the Blue-EGFP and Blue-DsRed vectors (Fig. 2E, F).
Generation of recombinant adenoviruses using the AdBlue system
Next, we generated adenoviruses expressing EGFP using the AdBlue system to test its efficiency. The AdBlue vector (linearized with ClaI) and the Blue-EGFP vector (linearized with EcoRI) were incubated together for 1 h in a PCR tube to facilitate enzymatic assembly (Fig. 3A; see Materials and Methods section). Then, the reaction products were transformed into cells. As predicted, blue colonies grew in LB–kanamycin agar. PacI digestion of the selected plasmids yielded the expected fragments (Fig. 3B). To verify the efficiency of the AdEasy system, plasmids extracted from 24 blue colonies were tested by PacI digestion, revealing 23 positive colonies (Fig. 3C). Then, the positive plasmids were digested with PacI to expose the inverted terminal repeats and transfected into AD-293 cells for virus packaging. The adenovirus genome vector was constructed using the AdEasy system as the positive control. As shown in Figure 3D, green fluorescence was detected 24 h after transfection, indicating that the foreign gene delivered by the vector was expressed in the host cells. Adenovirus packaging was confirmed by the appearance of green fluorescent plaques 5 days after transfection.

Generation of recombinant adenoviruses using the AdBlue system.
Modification of the AdBlue system to facilitate the generation of adenoviruses expressing CRISPR/Cas9 nuclease complexes
To expand its range of application, we modified the AdBlue system to simplify the generation of adenoviruses for the delivery of CRISPR/Cas9 nuclease complexes. Since the CRISPR/Cas9 system requires both a specific guiding sgRNA and Cas9 nuclease to modify the target gene, we constructed the Blue-Cas9 vector, to achieve expression of Cas9 nuclease, and the Blue-sgRNA vector. We developed the Blue-Cas9 vector to express Cas9 and DsRed (the two cassettes were separated by the 2A sequence) (Fig. 4A). Then, adenoviruses expressing Cas9 and DsRed (named Ad-Cas9) were generated using the AdBlue system. As predicted, after Ad-Cas9 infection, red fluorescence was detected in AD-293 cells (Fig. 4B), and Cas9 expression was verified by reverse transcription PCR (Fig. 4C). To differentiate it from the red fluorescence of the Blue-Cas9 vector, we constructed a Blue-sgRNA vector based on the Blue-EGFP vector. The sgRNA cassette was cloned from pLenti-sgRNA-Lib and was ligated into the Blue-EGFP vector using KpnI and XhoI sites(Fig. 4D). The Golden Gate cloning method was used to replace the ccdB cassette with annealed oligonucleotide pairs, as verified by endonuclease digestion (Fig. 4E). Then, adenoviruses expressing sgRNAs were generated with the AdBlue system. The viruses were used to infect AD-293 cells, and green fluorescence was detected, confirming successful virus packaging (Fig. 4F).

Rapid generation of recombinant adenoviruses expressing CRISPR/Cas9 nuclease complexes.
Functional testing of adenovirus-mediated target gene knockout efficiency in H9C2 cells
Finally, we assessed the target gene knockout efficiency induced by adenoviral delivery of CRISPR/Cas9 nuclease complexes in cultured H9C2 cells (Fig. 5A). We chose Celf1 as the target gene as it has been well studied in our laboratory. 18 The sgRNA binding site is in the sixth exon of Celf1 (Fig. 5B). Seven days after adenovirus infection, we extracted the genome of the cells as templates, amplified the sequence (∼1000 bp) around the target site by PCR, and subjected the fragment to sanger sequencing.19,20 DNA disruption was observed downstream of the predicted cleavage site in the Cas9 and sgRNA adenovirus-infected group (Fig. 5C, D). In this group, insertion–deletion mutations were detected using the T7E1 nuclease assay. No DNA disruption was observed in the control group by sequencing or T7E1 assay (Fig. 5D). Furthermore, we examined the Celf1 protein expression level by Western blot, and as predicted, there was significantly lower Celf1 expression in the Cas9 and sgRNA adenovirus-infected group (Fig. 5E, F).
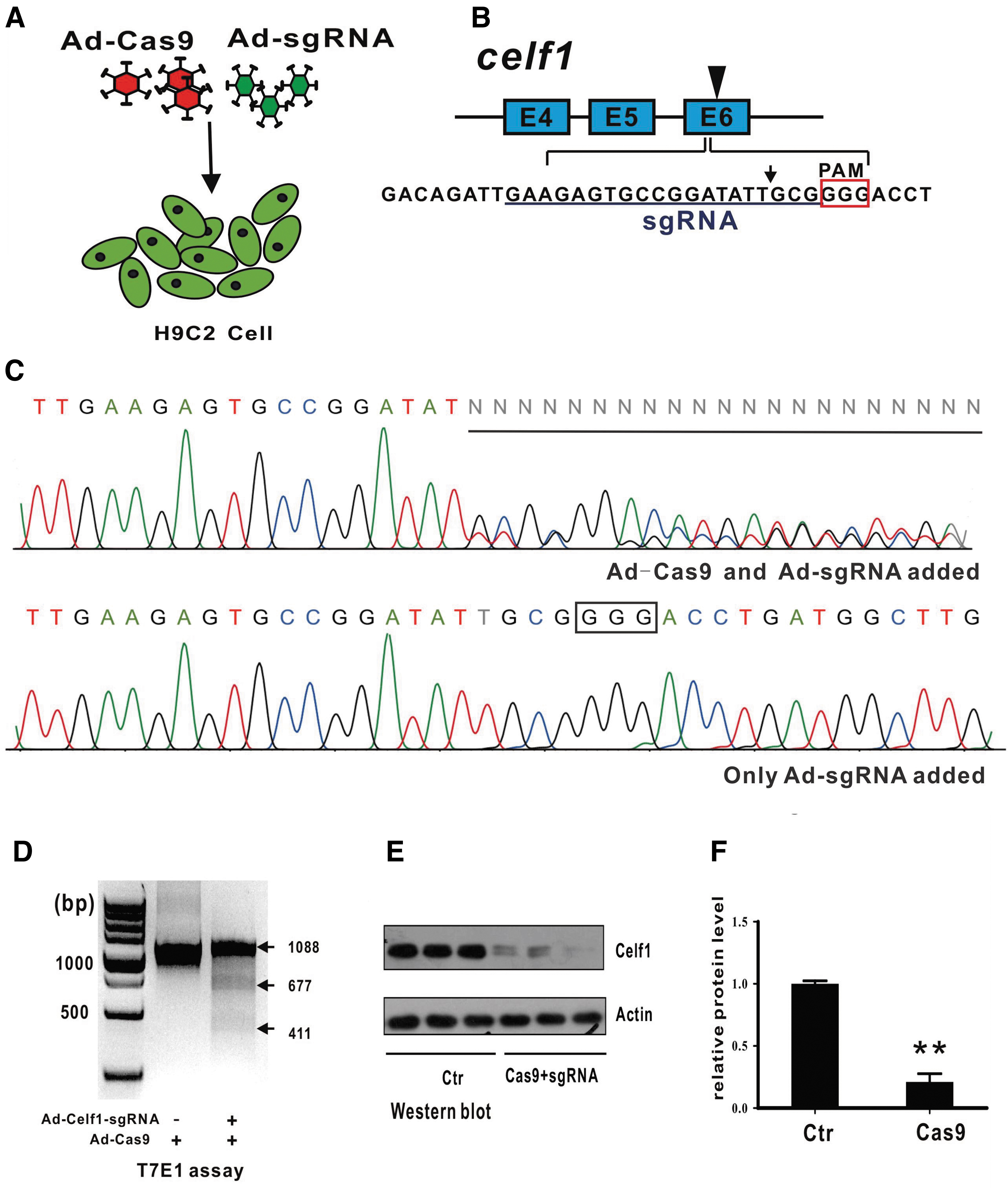
Functional testing of adenovirus-mediated target gene knockout efficiency in H9C2 cells.
Discussion
We developed a system based on enzymatic assembly to facilitate the efficient construction of adenoviruses. In our novel system, the GOI is cloned into the Blue vector, then the Blue and AdBlue vectors are linearized and ligated into the adenoviral plasmid in a one-step enzymatic reaction.1,12,21 Compared with conventional construction methods, the AdBlue system saves time and labor, which allows for high-throughput adenovirus generation.
The enzymatic assembly used in the AdBlue system was invented by Gibson et al. It has been rapidly adopted by synthetic biology laboratories because of its ease of use, flexibility, and suitability for large DNA constructs.16,17,22,23 We used this method to insert the GOI in the adenovirus genome. With this method, it is feasible to modify the adenovirus genome or even chemically synthesize the entire adenovirus genome, enabling the creation of tailored adenoviruses with distinct features.4,24–26 By replacing the construction of the adenoviral skeleton plasmid vector in the AdEasy system with enzymatic assembly, we ensured that the AdBlue system is rapid. In addition, the kanamycin resistance screening in the AdEasy system is inefficient for screening positive transformants, and at least 10–20 small colonies must be selected for analysis. Instead, we introduced blue–white screening to facilitate selection. Thus, positive transformants can be characterized by color and kanamycin resistance.
Adenovirus-mediated gene expression is short term because adenoviruses do not integrate into the genome of host cells, which limits the application of adenoviruses as vectors for gene therapy.4,27 Recently, CRISPR/Cas9 nuclease complexes have been widely used for gene editing. Short-term expression of the CRISPR/Cas9 complex in target cells can cause gene modification. However, if Cas9 is present for an extended period of time, it is associated with an increasing risk of DNA damage.5,6 Thus, based on the duration of gene expression, adenovirus particles have great promise as agents for CRISPR/Cas9 complex gene delivery. Indeed, there have been reports of efficient gene editing using adenoviruses as vectors.7,9 Several sgRNAs are typically designed to target different sites in a single gene, and the generation of the adenoviruses to express those RNAs is labor intensive. We modified the AdBlue system to simplify the generation of adenoviruses for the delivery of Cas9 and sgRNAs, and verified the efficiency of the simple system. In contrast, adenovirus-based Crispr/Cas9 delivery system can be divided into the unified system and separated vector system according to whether Cas9 and sgRNA are binding or not. A comparative study indicated that the functional virus titer of separate adenoviral system is ∼100 times than unified system. 28 Although the unified system is time and labor saving, the expression of Ca9 nuclease may affect the mutation efficiency mediated by the specific sgRNA. 29 Hence, we adopted separated adenovirus system to deliver Cas9 and sgRNA in our study.
There are three main advantages of the AdBlue system. First, in vitro adenoviral genome assembly occurs in a one-step reaction. Commonly used methods of adenoviral genome generation employ the efficient homologous recombination machinery of E. coli, which takes at least 2 days to complete and requires multiple steps.1,12,21 By contrast, adenovirus genome generation is a single-step 1-h process with the AdBlue system. The second advantage of the AdBlue system is the efficiency of positive colony selection. Both kanamycin resistance and blue–white selection enable accurate selection of positive colonies. Third, the vectors in the new system are derived from the AdEasy system, and the materials needed for the AdBlue system are common and commercially available. Therefore, the AdBlue system can easily be established in laboratories.
There are also limitations to the AdBlue system. The adenoviruses generated by the system are human adenovirus serotype 5 with deleted E1 and E3 genes, as in the AdEasy system. Thus, an immune response may be induced by residual virus gene expression, which limits the application of adenoviruses. 30 Another restriction is the packaging capacity of the adenoviruses generated by the AdBlue system. Large fragments (>8 kb) cannot be inserted into the adenovirus genome.
Conclusions
In summary, we described a novel system for recombinant adenovirus construction based on enzymatic assembly and the AdEasy system. The novel AdBlue system can markedly reduce the time and labor involved in adenovirus generation. To expand its range of application, we modified the AdBlue system to facilitate the generation of adenoviruses for the delivery of the CRISPR/Cas9 complex and successfully induced robust site-specific mutagenesis using such adenoviruses.
Footnotes
Authors' Contributions
Y.B., L.G., and G.J. conceived and designed the experiments. Y.B., L.G., J.W., Y.C., M.J., and Y.M. performed the experiments and contributed reagents/materials/analysis tools. Y.B., H.W., and G.J. wrote the article.
Acknowledgment
We thank Prof. Wensheng Wei for donating CRISPR/Cas9-associated plasmids.
Material Sharing Statement
To facilitate the spread of the novel adenovirus packaging system, we hereby declare to share all the plasmids involved in this study. Also, the plasmid profiles of pAd-Blue and pBlue are available in the Supplementary Data.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the National Key Research and Development Project (2019YFA0110400 to G.J.) and the National Foundation of Sciences and Technology (31971051, 31771562 to G.J.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.