
Abstract
Mutations in the human β-globin gene are the cause of β-hemoglobinopathies, one of the most common inherited single-gene blood disorders in the world. Novel therapeutic approaches are based on lentiviral vectors (LVs) or CRISPR-Cas9-mediated gene disruption to express adult hemoglobin (HbA), or to reactivate the completely functional fetal hemoglobin, respectively. Nonetheless, LVs present a risk of insertional mutagenesis, while gene-disrupting transcription factors (BCL11A, KLF1) involved in the fetal-to-adult hemoglobin switch might generate dysregulation of other cellular processes. Therefore, universal gene addition/correction approaches combining CRISPR-Cas9 and homology directed repair (HDR) by delivering a DNA repair template through adeno-associated virus could mitigate the limitations of both lentiviral gene transfer and gene disruption strategies, ensuring targeted integration and controlled transgene expression. In this study, we attained high rates of gene addition (up to 12%) and gene correction (up to 38%) in hematopoietic stem and progenitor cells from healthy donors without any cell sorting/enrichment or the application of HDR enhancers. Furthermore, these approaches were tested in heterozygous (β0/β+) and homozygous (β0/β0, β+/β+) β-thalassemia patients, achieving a significant increase in HbA and demonstrating the universal therapeutic potential of this study for the treatment of β-hemoglobinopathies.
Introduction
Sickle cell disease (SCD) and β-thalassemia belong to the group of β-hemoglobinopathies—autosomal recessive inherited blood disorders caused by abnormalities in the adult hemoglobin (HbA) and characterized by hemolytic anemia. In general, β-hemoglobinopathies are the most common genetic diseases in which a single gene is mutated. 1 To date, more than 300 mutations have been described in the β-globin (HBB) gene,2,3 affecting approximately 56,000 newborns with β-thalassemia and 270,000 newborns with SCD worldwide. 4 Mutations in the HBB gene lead to absent (β0) or reduced (β+) expression of HBB in patients with β-thalassemia, whereas a mutation at codon 6 (glutamine → valine) results in a structurally altered protein, causing SCD. Since β-thalassemia is an autosomal recessive disorder, only homozygous carriers suffer from clinical symptoms and exhibit either a mild form (thalassemia intermedia, β+β+ or β+β0) or severe form (thalassemia major, β0β0). 5 The impaired expression of HBB causes compensatory upregulation of α-globin, resulting in intracellular aggregation of hemoglobin. As a consequence, erythropoiesis is negatively affected by induced apoptosis of red blood cells and anemia. 6 Symptoms of these diseases include fatigue, headaches, and dizziness, which later evolve into various skeletal deformities and hepatosplenomegaly due to disturbed erythropoiesis and extramedullary blood formation. 7 Since thalassemia major patients present with pronounced anemia, lifelong blood transfusions are required, which are associated with immunosuppression, immunological reactions, and iron overload (hemosiderosis). 8 Therefore, the life expectancy of these patients is significantly reduced, with organ dysfunction being the main cause of death.
In the last decades, hematopoietic stem cell transplantation (HSCT) using allogeneic bone marrow, umbilical cord blood, or mobilized peripheral blood has shown remarkable success as an effective cure for β-thalassemia. 9 However, allogeneic HSCT is gravely limited by the scarce availability of matched sibling donors, and it is often associated with immunological complications, including graft rejection and graft-versus-host disease. 10 Therefore, autologous HSCT along with gene therapy tools are alternatives to allogeneic HSCT due to their lifelong treatment capacity. 11 Gene therapy approaches mainly aim to transfer a healthy copy of HBB or reinduce the expression of γ-globin and thus fetal hemoglobin (HbF). 12 Patients with co-inherited β-thalassemia and hereditary persistence of HbF are asymptomatic for the disease, demonstrating the therapeutic value of HbF as a treatment of β-hemoglobinopathies. 13 Currently, there are several ongoing gene therapy clinical trials for the treatment of β-hemoglobinopathies, including lentiviral gene transfer of a wild-type HBB gene (NCT01639690, NCT02151526, and NCT02453477) or γ-globin gene (NCT02186418), and CRISPR-Cas9 gene disruption approaches by means of non-homologous end joining (NHEJ) targeting the HBB repressor BCL11A to resurge the completely functional HbF (NCT03655678, NCT03745287, and NCT03282656).11,14 Though promising results have been observed for these gene therapies, a universal approach relying on CRISPR-Cas9 gene addition/correction by homology directed repair (HDR) could minimize the possible limitations of gene transfer and gene disruption strategies, avoiding semi-random integration of the lentiviral transgene or gene disruption of essential transcription factors involved in further cellular processes, respectively. 14
A universal gene correction approach ensures controlled targeted transgene integration and expression, blocking the transcription of the mutated HBB and enabling correction of all types of HBB mutations. Since HDR induction and the delivery of large constructs in primary cells is one of the major challenges in gene therapy, several research groups have focused on the correction by single-stranded oligodeoxynucleotide (ssODN) donor delivery of the most frequent single point mutations such as SCD (c.20 A > T) and IVS-I-110, attaining decent gene correction rates ranging from 7% to 25%.15,16 Alternatively, Dever et al. showed the potential of a universal gene correction in human hematopoietic stem and progenitor cells (HSPCs), attaining 19% of HDR rates. 17 In our study, we demonstrated high gene correction rates in HSPCs through two universal gene therapy strategies: gene addition of a cDNA containing untranslated and coding regions (untranslated region and coding sequence) of HBB, and gene correction through the delivery of a full-length corrected HBB copy. Both donor templates were provided by adeno-associated virus serotype 6 (AAV6) after targeting the promoter region (BetaPr) or the intron 1 (IVS-I) by CRISPR-Cas9 system.
Methods
Ethics statement
Human mobilized peripheral blood CD34+ HSPCs from individual donors were obtained using protocols approved by the local ethics committee/Institutional Review Board and after informed written consent (ethics number: 829/2016BO2), as well as a leukapheresis from Key Biologics (Memphis, TN). All methods were carried out in accordance with relevant guidelines and regulations.
Cloning
Oligonucleotides listed in Table 1 were cloned into pX330 plasmid (Addgene #42230) for further electroporation into the K-562 cell line, as previously described. 18 AAV6 donor template constructs were generated from the AAV6-GFP expressing construct pAAV.CMV.PI.EGFP.WPRE.bGH (Penn Vector Core, Pennsylvania, PA). Restriction sites for XhoI and NheI were included in the BetaPr, BetaPr DsRed, IVS-I, and IVS-I DsRed repair templates to be further cloned into pAAV.CMV.PI.EGFP.WPRE.bGH. Repair template sequences can be found in Supplementary Table S1. The resulting recombinant plasmids were named as pAAV-BetaPr, pAAV-BetaPrDsRed, pAAV-IVS-I, and pAAV-IVS-I DsRed. BetaPr and BetaPr DsRed donor template constructs were in vitro synthesized by GeneArt (Thermo Fisher Scientific, Waltham, MA), whereas IVS-I and IVS-I DsRed were purchased from Integrated DNA Technologies (IDT; Coralville, IA) and BioCat (Heidelberg, Germany), respectively. For BetaPr and IVS-I repair templates, we included a modified protospacer adjacent motif (PAM; BetaPr: AGG to ACG; IVS-I: TCC to TCA) with one nucleotide base pair change to avoid recutting by the CRISPR-Cas9 system. All plasmids were verified by Sanger sequencing.
sgRNA, single-guide RNA.
Production and concentration of AAV6
Two days before transfection, 5 × 106 Lenti-X 293T cells were seeded in a T175 culture flask (Corning, Corning, NY) without antibiotics. After 48 h, Dulbecco's modified Eagle's medium (DMEM; Biochrom, Cambridge, United Kingdom) was replaced by 9 mL Opti-MEM (Thermo Fisher Scientific). The cells were then transfected using JetPEI (Polyplus transfection) with 31.85 μg pAAV2.6 (Penn Vector Core, University of Pennsylvania, Philadelphia, PA), pAdDeltaF6 (Penn Vector Core), and transfer plasmid. After 6 h, cells were washed once with phosphate-buffered saline, and subsequently cultured in 12 mL DMEM supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY), 1% L-Glu (Biochrom), and 1% Pen-Strep (Biochrom). The viral supernatant was harvested 72 h post transfection, and the cellular debris was removed by centrifugation at 500 g. Afterwards, the virus was concentrated by ultracentrifugation (4°C/4 h/280,000 g) and resuspended in StemMACS™ HSC Expansion Media (Miltenyi Biotec, Bergisch Gladbach, Germany). Virus stocks were aliquoted and stored at −80°C.
AAV titration
To eliminate free viral DNA from disrupted capsids, 2 μL concentrated viruses were digested with 2 IU DNase I (New England Biolabs, Ipswich, MA) at a final volume of 40 μL. Samples were then incubated at 37°C for 30 min and at 75°C for 15 min. Next, 5 μL from the previous reaction were mixed with 1 μL proteinase K (Qiagen, Valencia, CA) at a final volume of 20 μL and incubated at 50°C for 30 min, following by 10 min at 98°C. Virus titer was calculated by quantitative polymerase chain reaction (qPCR) and droplet digital PCR (ddPCR) using the probe and primers listed in Table 2. The CFX Real-Time PCR (Bio-Rad Laboratories, Hercules, CA) was used to run the qPCR under the following conditions: 3 min at 95°C, 40 cycles consisting of 3 s at 95°C and 20 s at 60°C. For titer calculations by qPCR, a plasmid containing AAV inverted terminal repeat regions was used as an external standard. On the other hand, the ddPCR was run on the C1000 Touch Thermal Cycler (Bio-Rad) with the following thermal parameters: 10 min at 95°C, 40 cycles comprising 30 s at 95°C, 1 min at 57°C, and 2 min at 72°C, followed by enzyme inactivation at 98°C for 10 min.
PCR, polymerase chain reaction; qPCR, quantitative PCR; ddPCR, droplet digital PCR; PAM, protospacer adjacent motif; wt, wild type; HDR, homology directed repair.
CD34+ HSPC isolation
Peripheral blood was collected from donors after CD34+ HSPC mobilization. Subsequently, cell separation by Ficoll was performed to isolate peripheral blood mononuclear cells. Then, CD34+ HSPCs were positively selected following the manufacturer's instructions for CD34 MicroBead Kit UltraPure human or CliniMACS CD34 Reagent (Miltenyi Biotec), and MS separation columns (Miltenyi Biotec). Finally, cell purity was determined by flow cytometry analysis using the following antibodies (Miltenyi Biotec): CD34-APC, CD45-FITC, CD3-FITC, and CD14-PerCP. In all experiments, >95% of double-positive CD34+ CD45+ cells were obtained.
Gene editing of human CD34+ HSPCs
Two days prior to electroporation, CD34+ HSPCs were cultured in StemMACS™ HSC Expansion Media supplemented with 100 ng/μL stem cell factor (SCF), 20 ng/μL TPO, and 100 ng/μL Ftl-3 (Miltenyi Biotec). After 48 h, BetaPr or IVS-I single-guide RNAs (sgRNAs; IDT; Table 1) and Cas9 ribonucleoprotein (RNP; IDT) were mixed in a molar ratio of 1:2 (45–90 pmol) at room temperature for 15 min. Immediately afterwards, 1 × 105 CD34+ HSPCs were transfected using the 10 μL Neon transfection kit (Thermo Fisher Scientific) with the following electroporation settings: 1650 V, 10 ms, and three pulses. HSPCs were then transferred to differentiation media consisting of StemMACS™ HSC Expansion Media, 1% Pen-Strep, 1%
Gene editing analysis
CD34+ HSPCs were differentiated for 21 days into erythroid precursors, as previously described.18,19 On day 5, cells were collected for cell counting, DNA isolation, Sanger sequencing, and ICE analysis. On day 21, erythrocyte precursors were harvested for erythroid differentiation analysis and high-performance liquid chromatography (HPLC)-HbA quantification as performed before.18,19 Finally, HDR rates were quantified by a multiplex in-out ddPCR using the primers and probes listed in Table 2. B2M was used as a reference gene. To detect HDR rates for the different repair templates by ddPCR, in-out PCRs were designed, except for IVS-I, where in-out PCR is not feasible. Therefore, we designed a competitive ddPCR assay using an unlabeled wild-type probe in combination with a FAM-labeled probe for the modified PAM sequence of the IVS-I repair template.
To avoid detection of AAV ssDNA, isolated DNA was digested prior to ddPCR by S1 Nuclease (Thermo Fisher Scientific) using 1 μg total DNA, 4 μL 5 × reaction buffer, and 10 IU S1 in a total volume of 20 μL. Samples were incubated overnight at room temperature, and subsequently DNA clean-up was performed following the manufacturer's protocol for the QIAquick PCR Purification Kit (Qiagen). HDR was also determined by flow cytometry when using DsRed constructs. Cell recovery was determined by counting the number of cells at day 7 for each treatment using Trypan blue staining.
Off-target analysis
GUIDE-seq was performed and analyzed as previously described18,20 using 25 pmol dsODN and two different Cas9 endonucleases (IDT): SpCas9 wild type and SpHiFi Cas9. Off-targets were then double-checked by PCR, Sanger sequencing, and ICE analysis using the primers listed in Table 2.
Results
CRISPR-Cas9 design for targeting HBB locus
First, we selected five sgRNAs reported in earlier publications15,17,18,21,22 and screened them in K-562 cells in order to select the best two sgRNA candidates for further experiments in HSPCs. sgRNA-1 was designed to target the HBB promoter region, and sgRNA-2 the terminal sequence of exon-1, while both sgRNA-3 and sgRNA-4 target intron-1 (IVS-I), and sgRNA-5 targets intron-2 (IVS-II; Fig. 1A). All sgRNA-encoding oligo pairs were annealed and ligated into a pX330 vector and subsequently transfected in K-562. T7E1 and ICE analysis on day 5 revealed that sgRNA-1 and sgRNA-3 achieved the highest rate of insertion and deletions (indels; sgRNA-1 T7E1: 39.6 ± 8.4; ICE: 59 ± 17.8; sgRNA-3 T7E1: 38.6 ± 8.1; ICE: 47.7 ± 8.1). Even though the rest of the sgRNAs attained decent gene editing scores (Fig. 1B) by T7E1 (sgRNA-2: 28.5 ± 6.4; sgRNA-4: 23.4 ± 6.9; sgRNA-5: 28 ± 0.4) and ICE analysis (sgRNA-2: 16 ± 3.5; sgRNA-4: 7.3 ± 1.5; sgRNA-5: 11.3 ± 3.5), only the two best candidates were selected (sgRNA-1 and sgRNA-3) for further experiments. Due to the superior efficacy and low toxicity of chemically modified sgRNA over unmodified sgRNAs, 23 our finalized candidates—sgRNA-1 (BetaPr) and sgRNA-3 (IVS-I)—were chemically synthesized with three terminal modifications. HSPCs were then electroporated after sgRNA/Cas9 RNP complex generation, obtaining high levels of gene editing by ICE analysis (sgRNA-1: 84.25 ± 5.7; sgRNA-3: 87.43 ± 6.9; Fig. 1B).

Gene addition approach in healthy CD34+ HSPCs.
HBB gene correction repair template design and assessment in healthy HSPCs
For gene correction experiments, we designed two different repair templates based on the sgRNA targeting position. In the case of BetaPr (sgRNA-1), which targets the HBB promoter region, we proposed a transgene addition strategy through the delivery of a mutation-free HBB cDNA containing the minimal part of the HBB promoter with the corresponding UTRs. For easy assessment of transgene addition, we designed an additional donor template with a red fluorescent marker (DsRed) instead of HBB cDNA (Fig. 1C). Both donor templates were subcloned into gene transfer plasmid for AAV6 virus production and verified by sequencing (data not shown). Viruses were produced, and titers were calculated by qPCR and ddPCR using the primers listed in Table 2. For AAV BetaPr, a mean value of 9.2 × 1013 and 8.1 × 109 TU/mL were determined by qPCR and ddPCR, respectively, while for AAV BetaPr DsRed, the virus titer was 1.2 × 1014 and 7 × 109 (Fig. 1D). Next, we assessed the gene addition capacity of AAV6 donor templates at different MOIs (2,000, 1,000, 500, and 250) in HSPCs cultured in erythroid differentiation media immediately after gene editing. The indicated MOIs were determined by ddPCR due to its precise and absolute quantification without the need for any external standard. Consequently, our MOIs based on ddPCR were lower in comparison to MOI calculations in previous studies using the qPCR method.
At day 14 post treatment, cells transduced with the DsRed constructs were harvested for flow cytometry analysis and DNA isolation for subsequent transgene correction assessment by ddPCR. Our results demonstrated that both methods showed a similar level of transgene correction (FACS: 8.8 ± 1%; ddPCR: 7.3 ± 0.95%), where AAV6 MOI of 1,000 exhibited the best integration score (Fig. 1E and F). Similar HDR efficiencies were observed for BetaPr construct with a MOI of 1,000 (ddPCR: 8.1 ± 0.7%; Fig. 1G). In addition, cells were counted at day 7 for each treatment to assess cell recovery after CRISPR-Cas9 transfection and AAV transduction. In this regard, cell recovery rates were higher for lower MOIs and AAV treatment in comparison to CRISPR + AAV (Fig. 1H). Nonetheless, cell viability of surviving cells was comparable to the control and was not impaired during the erythroid differentiation process (Fig. 1I).
For IVS-I (sgRNA-3), we aimed to correct the entire HBB gene by providing a healthy copy with a PAM variation to avoid recutting by the CRISPR-Cas9 system. Furthermore, a comparable repair template was designed carrying the DsRed gene separated by T2A sequence, where the expression of DsRed is controlled by the endogenous HBB promoter after successful integration (Fig. 2A). Similar to our earlier design, both constructs were cloned in AAV6 transfer vector, and their viral titer were determined by qPCR and ddPCR (IVS-I qPCR: 4.6 × 1013 IU/mL; IVS-I ddPCR: 1.6 × 1010 IU/mL; IVS-I DsRed qPCR: 4.2 × 1013 IU/mL; IVS-I DsRed ddPCR: 9.5 × 109 IU/mL; Fig. 2B). We conducted a screening for selecting the optimal MOI for our AAV donor and found that a MOI of 2,000 increased HDR (IVS-I DsRed FACS: 24.9 ± 4.7%; ddPCR: 26.13 ± 0.7%; IVS-I ddPCR: 30.83 ± 5%; Fig. 2C–E). Finally, as observed for BetaPr constructs, cell recovery was hindered during the first days in AAV- and CRISPR + AAV-treated samples (Fig. 2F). However, cell viability was similarly high as the control throughout the erythroid differentiation (Fig. 2G).
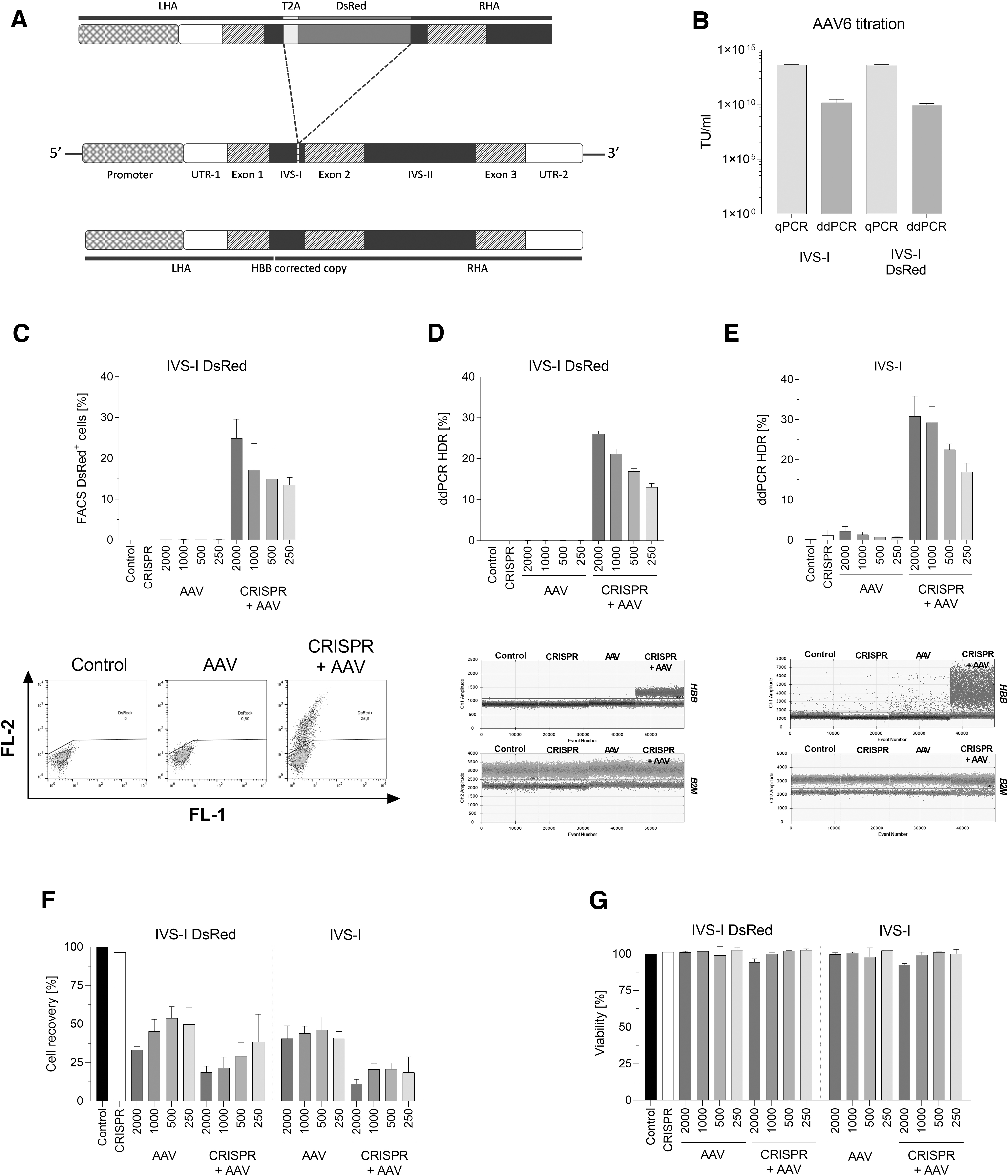
Gene correction approach in healthy CD34+ HSPCs.
Gene editing of HSPCs from a β8-88/IVS-I-110 G > A heterozygous patient (β0/β+)
Since our gene correction data with healthy HSPCs showed efficient gene addition and correction, we extended our analysis to HSPCs from a heterozygous β-thalassemia patient (β0/β+). Our gene editing experiment using these patient cells showed up to 70.3% and 78% indel rates for BetaPr and IVS-I, respectively. For the optimal MOI, we relied upon earlier results where MOIs of 1,000 and 500 were used for gene addition using BetaPr sgRNA, and MOIs of 2,000 and 1,000 were used for gene correction using IVS-I sgRNA. As observed in healthy HSPCs experiments, BetaPr and IVS-I constructs achieved similar results in comparison to the DsRed constructs, spotting the best HDR rates with MOIs of 1,000 and 2,000, respectively (Fig. 3A–F). Remarkably, since this patient is heterozygous (β8-88/IVS-I-110 G > A) and one allele has a mutation in the target site of the IVS-I sgRNA-3, gene correction results achieved in this experiment represented the half attained in healthy HSPCs (see Fig. 3F and Fig. 2E, respectively). The BetaPr DsRed strategy targeting HBB promoter showed HDR rates of 3.3 ± 0.1% and 3.1 ± 0.2% by flow cytometry and ddPCR, respectively (Fig. 3A and B), whereas the IVS DsRed strategy targeting the IVS-I region accomplished 8.6 ± 0.7% and 10.7 ± 1.0% of HDR by flow cytometry and ddPCR, respectively (Fig. 3D and E). More importantly, BetaPr and IVS-I approaches achieved a mean value of 4.2 ± 1.0% for gene addition and 16.6 ± 1.7% for gene correction (Fig. 3C and F). The cell recovery was lower in CRISPR and AAV treatments in comparison to the control (Fig. 3G). However, cell viability was not affected at day 14 (Fig. 3H). Interestingly, even though gene editing frequencies were lower in comparison to healthy HSPCs, HPLC revealed that these gene correction efficiencies were sufficient to induce HbA production (Fig. 3I). In comparison to AAV-treated samples, we could achieve 2.3-fold (MOI 2,000) and 4.5-fold (MOI 1,000) HbA increases with IVS-I, whereas 2.3-fold (MOI 1,000) and 1.4-fold (MOI 500) HbA increases were attained with BetaPr in CRISPR and AAV treatments.
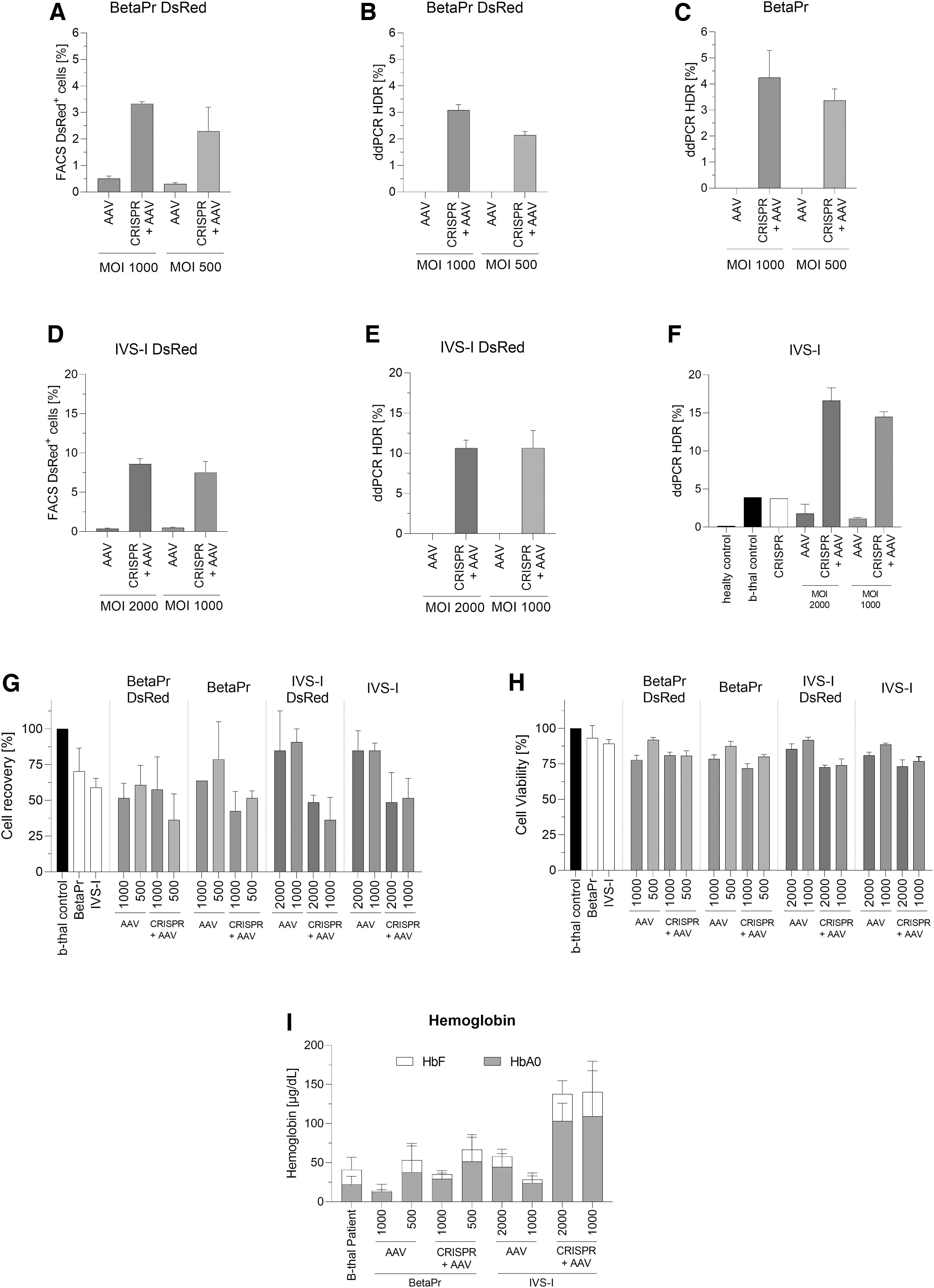
Gene editing of HSPCs from a β8-88/IVS-I-110 G > A heterozygous patient (β0/β+).
Gene editing of HSPCs from an IVS-I-1 homozygous patient (β0/β0)
After performing gene correction in a heterozygous patient (β0/β+) with reduced expression levels of HbA, we applied our approaches to HSPCs obtained from a thalassemia major patient (β0/β0, IVS-I-1 G > A) with no HbA production. Particularly, this patient expresses HbF to compensate for the absence of HbA, but the fetal levels are not sufficient to be completely therapeutic. ICE analysis after CRISPR treatment revealed 65% and 96% indel rates for BetaPr and IVS-I sgRNAs, respectively. Regarding HDR rates, the BetaPr DsRed strategy achieved 5.68 ± 0.74% of integration determined by flow cytometry (Fig. 4A) and 3.29 ± 0.73% by ddPCR (Fig. 4B). When utilizing the BetaPr repair template, a similar HDR efficiency was attained (ddPCR: 6.39 ± 1%; Fig. 4C). Interestingly, higher gene correction rates were achieved by using the IVS-I-DsRed construct (Fig. 4D: flow cytometry 21.00 ± 0.17%; Fig. 4E: ddPCR 21.97 ± 1.76%) and IVS-I repair template (Fig. 4F: 12.18 ± 0.99%). As previously described, the combination of CRISPR electroporation and AAV transduction significantly reduced cell recovery (Fig. 4G). However, the cells did not lose their proliferation and differentiation capacity. Indeed, differentiated erythrocyte precursor cells at day 21 post treatment increased their HbA levels from 0.33 ± 0.09 μg/dL to 31.61 ± 10.77 μg/dL for BetaPr and from 0.40 ± 0.02 μg/dL to 64.99 ± 35.64 μg/dL for IVS-I in CRISPR + AAV cells in comparison to the AAV-treated samples (Fig. 4H).

Gene editing of HSPCs from an IVS-I-1 G > A homozygous patient (β0/β0).
Gene editing of HSPCs from an IVS-I-110 G > A homozygous patient (β+/β+)
Next, we tried to extend our portfolio to a homozygous IVS-I-110 patient (β+/β+), since we wanted to corroborate the universality of our IVS-I approach due to the existence of a HBB point mutation exactly in the target region of IVS-I sgRNA in this patient. CD34+ HSPCs were isolated and the two most efficacious AAV MOIs were selected for each strategy to transduce the cells. Since this patient carries the IVS-I-110 G > A mutation on both alleles, a variant of sgRNA-3 (IVS-I mut) was designed and tested. The HDR efficiencies for BetaPr and IVS-I were comparable to their respective DsRed counterparts, whereas the sgRNA IVS-I mut outperformed the wild-type version (Fig. 5A–F). ICE analysis for BetaPr and IVS-I revealed up to 49.6% and 14% of indels, respectively. Also, the indel rates attained for IVS-I mut were up to 53.2%, which elucidates the lower HDR rates achieved in this patient. Cell recovery was lower in CRISPR and AAV treatments in comparison to the control (Fig. 5G). Again, cell viability was not affected (Fig. 5H). Furthermore, despite low gene editing frequencies, HPLC analysis showed that the gene correction strategy with sgRNA IVS-I mut significantly induced resurgence of HbA (Fig. 5I). In comparison to AAV-treated samples, we could achieve 1.7-fold (MOI 2,000) and 1.9-fold (MOI 1,000) HbA increases with IVS-I mut sgRNA in CRISPR and AAV treatments. Importantly, CRISPR gene disruption with IVS-I and IVS-I mut sgRNAs also induced HbA expression.

Gene editing of HSPCs from an IVS-I-110 G > A homozygous patient (β+/β+).
Safety assessment of the gene editing strategies
Since BetaPr and IVS-I sgRNAs presented elevated numbers of in silico predicted off-targets (Table 3), GUIDE-seq was performed to determine the specificity and fidelity of these sgRNAs quantitatively. Moreover, in addition to wild-type Cas9 protein, we performed a head-to-head comparison with HiFi Cas9 endonuclease, considered to have higher specificity without changes in gene editing efficiency, which was confirmed in our results (Fig. 6A–E). For BetaPr sgRNA, 20 off-targets were detected using wild-type Cas9 (Fig. 6B), whereas only nine were identified when utilizing HiFi Cas9 (Fig. 6C). Regarding IVS-I sgRNA, 25 and 8 off-targets were obtained with wild-type Cas9 and HiFi Cas9, respectively (Fig. 6D and E). In addition, after a BLAST analysis of the off-target sequences, we identified matches to six genes (MYPT3, NCOR2, LGALS8, RPL23A, TTN, and TTC7A) for BetaPr sgRNA, whereas three genes (ASTN2, MIIP, and SYNE4) were found for IVS-I sgRNA, respectively. Nonetheless, the sequences were spotted in non-coding regions. Also, none of these off-targets were present within the HBD gene, despite the high degree of homology to the HBB sequence. To cross-verify the off-target sites, we selected all the off-targets we noted for HiFi Cas9-treated samples for both sgRNAs (Fig. 6C and E) and performed independent ICE analysis after Sanger sequencing. Our data demonstrated that no detectable double-stranded breaks (DSBs) were noted by ICE analysis for all the tested samples in their respective sites (Fig. 6F).
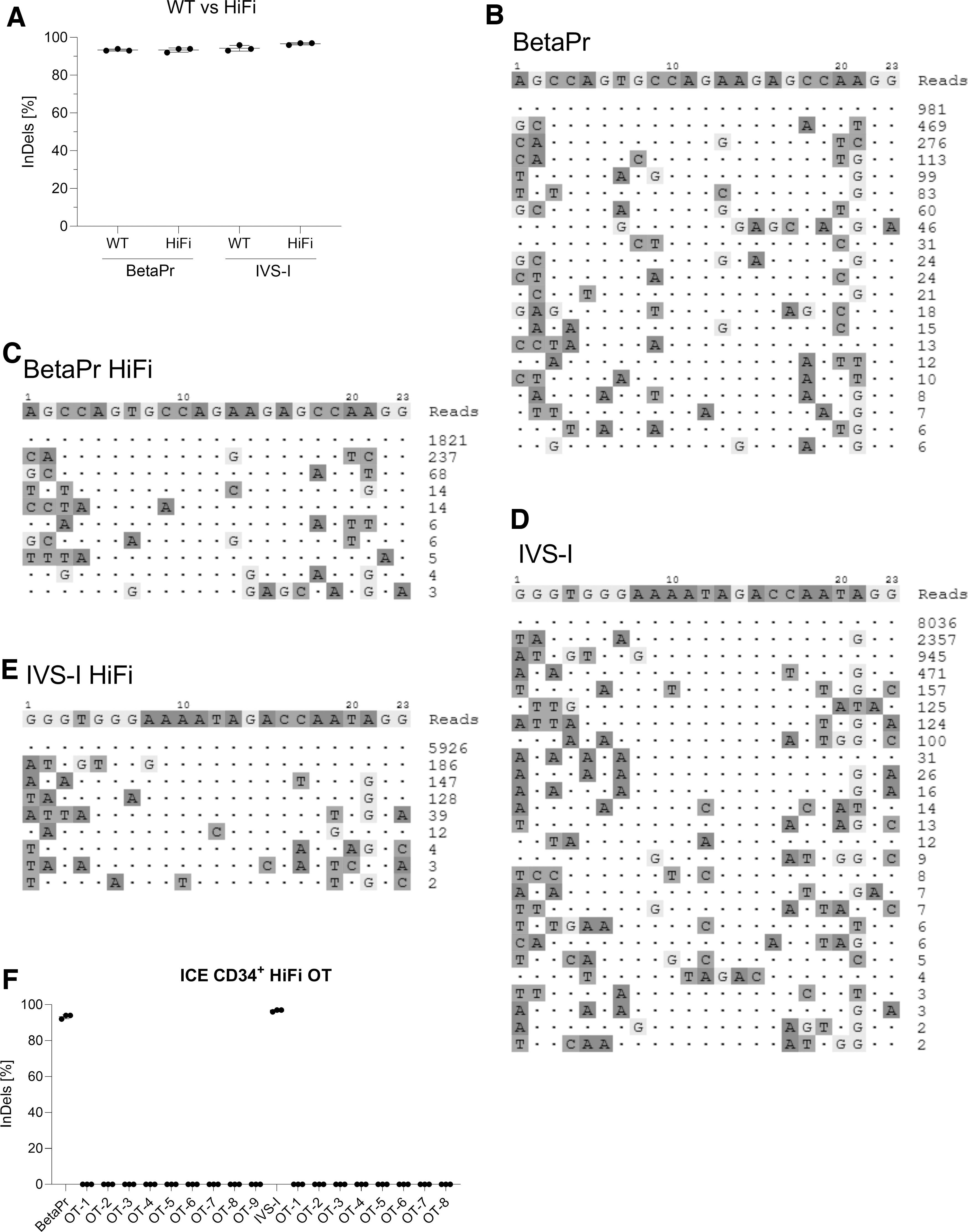
Safety assessment of the gene editing strategies.
Discussion
Lentiviral gene transfer and gene disruption for β-hemoglobinopathies have already reached the clinics with a successful outcome, while gene correction approaches are still at the preclinical stage due to the challenging induction of HDR events in primary cells. However, thanks to great advances in the field of gene editing, attaining gene correction efficiencies at therapeutic levels have been possible through the delivery of the repair template by ssODNs, integrase-deficient lentiviral vectors (IDLVs), and AAV.16,17,24–28 Although lentiviral gene transfer has exhibited safe and promising results, semi-random integration of the transgene close to oncogenes or suppressor genes might occur. Furthermore, since HBB needs a controlled expression by the locus control regions, large constructs are required to be delivered, which hinders the virus production and affects surrounding gene rearrangement after transgene insertion. Alternatively, induction of HbF by knocking down the expression of BCL11A, a transcription factor involved in γ- to β-globin switching, has demonstrated to be a more straightforward strategy in terms of safety and gene editing efficiency. 18 Nonetheless, BCL11A is an important transcription factor involved in hematopoiesis, and its disruption might negatively affect a patient's health several years post treatment, although no adverse effects have been reported to date. Indeed, six SCD patients have been treated with the BCH-BB694 lentiviral vector, which encodes an interference RNA that suppresses BCL11A expression in an erythroid lineage-specific manner. All patients safely engrafted the edited cells and upregulated their levels of HbF in the range of 20.4–41.5% after an average of 18 months of follow-up. 29 Likewise, the CLIMB THAL-111 and CLIMB SCD-121 clinical trials sponsored by CRISPR Therapeutics and Vertex Pharmaceuticals have provided splendid results as the first lentiviral free gene therapy for transfusion-dependent β-thalassemia and SCD using CRISPR-Cas9 targeting BCL11A enhancer (CTX001). Both patients (β0/β+ and βS/βS) increased their F-cells to almost 100%, and even though they presented adverse symptoms, none of them were considered to be related to the CTX001 infusion. Currently, the patients are transfusion independent after a follow-up of 15–18 months. 30 Unlike gene transfer and gene disruption strategies, gene correction has the advantage of simulating normal cell conditions without causing significant changes at a genomic level. Therefore, in the present study, we demonstrated the therapeutic potential of two universal gene correction approaches using CRISPR-Cas9 and AAV6 donor templates, which could be implemented in the clinic for the treatment of β-hemoglobinopathies regardless of the type of mutation in the HBB gene.
AAV6 was selected as donor template delivery platform in HSPCs due to its large carrying capacity, but also based on previously reported results showing its superiority over ssODN, IDLV, and adenovirus in terms of gene correction rates and genotoxicity. 31 Furthermore, in vivo experiments in xenograft mice were performed using HSPCs edited by CRISPR-Cas9 and AAV6, presenting decent cell engraftment capacity, though a reduction in gene-edited repopulated cells was observed. 17 In our study with healthy HSPCs, we achieved therapeutic levels of gene correction in HSPCs without any cell enrichment or the use of HDR enhancers. Furthermore, similar HDR efficiencies were obtained with constructs with or without reporter genes, revealing that gene correction might be locus dependent. Also, the gene correction frequencies attained in this study were comparable or higher than other publications utilizing ssODN, IDLV, or AAV6 as the repair template delivery method.15,16 However, previous research has mainly focused on the correction of SCD mutations, and thus repair templates did not cover the whole HBB gene, except for Hoban et al. who delivered a full HBB copy and a sgRNA under the influence U6 promoter by IDLV. 24 In addition, excellent gene correction rates were accomplished in our study when using HSPCs from β-thalassemia patients, even for the heterozygous patient, where HDR rates were half as expected, indicating that the second allele (with IVS-I-110 mutation) was targeted with lower efficiency. Nonetheless, cell recovery was found to be lower in the CRISPR + AAV-treated samples in comparison to the control. We presume that the noticed delay in proliferation and apoptosis might be majorly driven by activation of p53-mediated DNA damage response pathway in the CRISPR-Cas9 and AAV-treated group reported recently. 32 Furthermore, the variation in the toxicity and efficiency of gene editing may also depend on the AAV purification method (ultracentrifugation, cesium chloride, or iodixanol gradient), the heterogeneity of the HSPC population when enrichment is only performed for the CD34 marker, and the variation in expression of the receptors/co-receptors in this heterogenic population. 33 Therefore, the gene editing approach that we propose in this study could be further improved by addressing the above-listed challenges. Most importantly, even though we attained moderate gene correction/addition frequencies in the patient samples compared to the healthy HSPCs, gene editing efficiencies were sufficient to induce HbA upregulation with therapeutic potential, especially in the thalassemia major patient (β0/β0) where the efficacy is strongly noted. Moreover, as described in previous publications, residual NHEJ leads to HbF resurgence. Probably due to the inhibition of HBB expression, the cells try to compensate for the hemoglobin imbalance. 16
Once it had been determined that both universal strategies led to high gene correction efficiencies and HbA resurgence, we assessed the safety of these approaches by GUIDE-seq analysis. Moreover, a comparison between wild-type Cas9 and HiFi Cas9 endonuclease was performed, since earlier studies reported that HiFi Cas9 presents higher target specificity without changes in on-target efficiency.16,34 This fact was observed in our results. However, still nine and eight off-targets were detected for BetaPr and IVS-I sgRNAs, respectively. We assessed each of the off-targets in their respective genomic region and noted that none of these 17 off-targets falls in the protein coding region, and these genes are not enriched for any specific pathway. Moreover, the frequency of off-targets is relatively much lower compared to on-target DSBs (>85%) for both sgRNAs. Additional gene ICE analysis was performed to confirm these results, and no detectable DSBs were observed for those off-target sites in gene-edited HSPCs after gene amplification, sequencing, and ICE analysis. Nonetheless, further optimization such as truncated versions of sgRNA, optimized GC content, and DNA–RNA chimera might also increase the specificity of BetaPr and IVS-I sgRNAs. 24
Conclusions
Although a universal approach was reported by Cai et al., this strategy was performed in induced pluripotent stem cells after the excision of endogenous HBB using two sgRNAs. 35 Alternatively, in this study, we demonstrated a HbA resurgence in patient HSPCs after obtaining respectable gene correction rates for two universal approaches, in which the full HBB gene correction was the most promising approach. Moreover, safety analysis of our strategies revealed possible clinical translation. Nevertheless, further experiments are required to refine these approaches, such as optimization of sgRNA with no detectable off-targets, increasing cell viability by utilizing different AAV purification protocols, and reducing the variability between donors by performing gene correction in a concrete HSPC subpopulation. Also, HSPC engraftment experiments in a next-generation sequencing mouse model could be performed to determine the repopulation capacity of the gene-edited HSPCs. After all these analyses, the technology might be transferred to a clinical Good Manufacturing Practice device for further clinical application. It is worth noting that the proposed design of repair templates for gene addition and gene correction can be easily transferred to other genetic diseases and might help to develop similar therapeutic strategies in other fields.
Footnotes
Acknowledgments
The authors would like to express their gratitude to the Stem Cell Laboratory at the University Children's Hospital of Tübingen, for providing us CD34+ HSPCs. Also, we would like to thank the technical support regarding AAV ssDNA digestion from Malte Ritter, Jérémy Haaf, and Prof. Julia Skokowa from the Division of Translational Oncology, Department of Hematology, University Hospital Tübingen, Germany.
Author Disclosure Statement
None of the authors state any conflicts of interest.
Funding Information
This study was successful thanks to the financial support of the research funding programs Jürgen Manchot Stiftung, Fortüne Tübingen (no. 2412-0-0; no. 2485-0-0), Clinician Scientist Program (no. 440-0-0), Förderverein für krebskranke Kinder Tübingen eV, and the University Children's Hospital of Tübingen.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.