
Abstract
Over the past two decades, the discovery of CRISPR-Cas immune systems and the repurposing of their effector nucleases as biotechnological tools have revolutionized genome editing. The corresponding work has been captured by 90,000 authors representing 7,600 affiliations in 126 countries, who have published more than 19,000 papers spanning medicine, agriculture, and biotechnology. Here, we use tech mining and an integrated bibliometric and networks framework to investigate the CRISPR literature over three time periods. The analysis identified seminal papers, leading authors, influential journals, and rising applications and topics interconnected through collaborative networks. A core set of foundational topics gave rise to diverging avenues of research and applications, reflecting a bona fide disruptive emerging technology. This analysis illustrates how bibliometrics can identify key factors, decipher rising trends, and untangle emerging applications and technologies that dynamically shape a morphing field, and provides insights into the trajectory of genome editing.
Introduction
While genome editing has been on the rise over the past two decades, the advent of CRISPR-based technologies has accelerated and democratized genome editing in the past 9 years.1,2 Several Cas-based molecular machines have been co-opted from the bacterial adaptive immune system3 to generate CRISPR-based technologies, such as sgRNA:Cas9, 4 which have enabled facile genome editing since 2013.5,6 Recently, the leading developers of this genome editing technology were awarded the 2020 Nobel Prize in Chemistry, illustrating the tremendous potential and impact of this technology. Early work focused on deciphering the molecular processes that drive CRISPR-based adaptive immunity in bacteria 7 and the development of programmable Cas proteins that laid a preparatory foundation for CRISPR-based technologies. 8 Subsequently, these Cas effectors were deployed to manipulate genomes, transcriptomes, and epigenomes in a broad diversity of organisms across the tree of life, such as bacteria, plants, and humans. 9 More recently, these CRISPR-based technologies have been widely adopted to engineer model organisms and even develop gene therapies tested in clinical settings.10,11 Besides Cas9, the CRISPR toolbox has been expanded to encompass various Cas effector proteins such as Cas9, Cas12, Cas13, and the Cascade complex. 9 As tools continue to be optimized with regards to specificity, efficiency, and delivery modalities, the intellectual property (IP) landscape is being defined12–14 to enable widespread exploitation in medicine (e.g., gene therapies and antimicrobials), agriculture (e.g., crop breeding and disease resistance in livestock), and biotechnology (e.g., enzyme engineering and biofuel genesis). The accessibility and dissemination of CRISPR tools via repositories such as Addgene have allowed broad access to the best tools by academics and nonprofit organizations across the globe. 2
Though the rise of genome editing and global spread of CRISPR tools is undeniable, relatively little is known about the geographical, topical, individual, and collaborative patterns that drive this academic phenomenon and commercially disruptive technology. 15 Here, we implemented an integrated research framework, using a bibliometric approach,16,17 augmented by text mining, analysis of abstract record compilations, and a scientific evolutionary pathway (SEP) analysis,18,19 to investigate the underlying patterns that have driven the adoption and implementation of CRISPR technologies. Specifically, we analyzed publication trends and authorship patterns for the CRISPR and the genome editing literature over space and time using queries in Web of Science to identify key contributors and influential papers, as well as the topics and biases that have shaped and are currently driving the field.
Methods
Publication records were retrieved using text queries mining Web of Science records as of March 25, 2021, spanning manuscripts published between 2000 and 2020. Records were retrieved and cross-indexed using entries providing information with regards to manuscript authors, affiliated institutions, publication journal, year, title, and abstracts. For SEP analysis, we used the method pioneered by Zhang et al. 20 to trace the evolution of scientific topics into different subtopics by identifying a linguistic predecessor–descendant relationship from these bibliometric data. We then used the SEP approach to track the convergence and divergence of research topics on genome editing research and to discover potential connections between these topics within a knowledge flow.
Generally, we ascribed six definitions as follows:
Definition 1. An article is represented by a vector (article vector): its feature space consists of terms of the entire data set, and its cell represents the frequency of a given term appearing in this article.
Definition 2. A topic is a collection of articles sharing similar semantic content, a lexicon, and is geometrically represented as a circle, with a centroid measured by the mean of all involved article vectors, and a boundary measured by the largest Euclidean distance between the centroid and all other article vectors.
Definition 3. Articles published in the same year are organized in one time slice. The entire data set is analyzed as a bibliometric stream, that is, the SEP algorithm is to analyze each time slice sequentially according to the order of publication year, and for each time slice, the algorithm is to analyze each article sequentially according to the order of unified publication ID.
Definition 4. Initial topics are those consisting of articles in the first time slice and are starting points of the evolutionary pathways. Initial topics usually represent the root (e.g., original ideas and concepts) of the case (i.e., CRISPR in this paper).
Definition 5. A topic has two status categories, either “live” or “dead,” as defined by “sleeping beauties,” 21 for which a topic could “die” if it does not receive new articles in certain sequential time slices, and a “dead” topic could be revived and “alive” again if a newly born topic shares the highest similarity with it.
Definition 6. A community is a group of proximate topics in a network—usually a branch in a SEP map—which represents a subfield of the case.
Based on the above definitions, we implemented a stepwise algorithm to create the SEP as follows:
Step 1: All articles in the first time slice are grouped as one initial topic, which is set as the starting point of the evolutionary pathways. The algorithm moves to the second time slice and analyzes its involved articles one by one.
Step 2: We measure the cosine similarity between a current article and the centroids of all “live” topics.
Step 3: We assign the article to its most similar topic. If the Euclidean distance between the article and the centroid of the assigned topic is smaller than its boundary, this article will be directly involved in the topic. Otherwise, it will be labeled as “drift.” Then, we return to Step 2 and analyze the next article until the end of this time slice.
Step 4: After analyzing all articles in one time slice, we check the status of each topic, that is, set topics as “dead” if they meet with the constraint in Definition 4 (a parameter is used here to decide the length of sequential time slices). For each “live” topic, an unsupervised K-means approach is introduced to group its assigned “drift” articles into certain subtopics (an interval for seeking the local-optimal number of topics is required).
Step 5: We measure the cosine similarity between each subtopic and two sets of topics—its assigned “live” topic and all “dead” topics. If the most similar topic of the subtopic is its assigned one, their relationship is defined as “predecessor–descendent.” Otherwise, the most similar “dead” topic will be revived and set as “live” and then becomes the predecessor of the subtopic.
Step 6: We label a new topic (i.e., a subtopic in Step 5) via the term with the highest similarity with all other terms in the topic—if the term has already been used before, choose the term with the second highest similarity, et cetera.
Step 7: We update the centroid and boundary of all “live” topics, and the algorithm moves to the next time slice, and we return to Step 2.
Results of the SEP approach include a list of topics and their predecessor–descendant relationships. These topics are then visualized in a network via Gephi. 22 In the network, each topic is represented by a node, and the size of a node represents its importance, as measured by the value of term frequency inverse document frequency (tf-idf) analysis. A directed edge represents the predecessor–descendant relationship between its connected nodes, and the weight of an edge reveals the strength of the relationship (e.g., semantic similarity). The color of nodes reflects their communities identified by an approach of community detection integrated in Gephi as “modularity.” 23 Similarity measurements were carried out for the 119 topics identified across the three distinct time periods (9 topics pre-2013, 64 topics between 2013 and 2018, and 46 topics since 2019), using semantic similarity coefficients. Details are available at https://github.com/IntelligentBibliometrics/Gene-editing.
Results
CRISPR technology fueled the rise of the genome editing literature
To provide quantitative and qualitative insights into the drivers of the CRISPR craze, 24 we first defined the genome editing lexicon of interest and quantified relevant publications over the past 20 years, focusing on articles, reviews, and letters comprising 26,484 records (Supplementary Table S1). Results show that the CRISPR literature (more than 19,000 papers published since 2000 by 90,000 authors from around 7,600 institutions located in 126 countries; Supplementary Table S2) is rapidly growing, and that CRISPR-based tools impressively overtook incumbent technologies such as ZFNs, TALENs, and Meganucleases in 2013 (Fig. 1A), within months of publication of the first proof of concept for CRISPR-based genome editing in human cells.5,6 Currently, CRISPR-related publications account for the near totality of the genome editing field and are more than 10 times more numerous than ZFN, TALEN, and Meganuclease papers combined (Fig. 1A). Indeed, publications related to these first-generation genome editing technologies have been in decline since the advent of CRISPR-based genome editing technologies in 2012 (Fig. 1A).

Genome editing–related publications since 2000.
Remarkably, despite this rapid early adoption pattern, especially in the United States and China, the CRISPR literature continues to expand at an impressive rate (Fig. 1A), perhaps suggesting that genome editing is yet to hit maturity as a field, which is consistent with the continued dissemination of CRISPR tools across the planet.1,2 Importantly, this shows how CRISPR as a field evolved from a relatively small “niche” microbiology topic into the major driver of genome editing in 2013, establishing a “before CRISPR” era 25 and perhaps an “after displacement” of incumbent technologies period thereafter. This rise was fueled by the advent of the single-guide RNA technology in 2012, which quickly enabled genome editing (Fig. 1B) and prompted an explosion in genome editing studies and citations (Fig. 1C), as recognized by the 2020 Nobel Prize in Chemistry selection committee. Critical advances achieved in the past 2 years are also notable, with development of novel base editing tools and polished technologies such as prime editing,26,27 as well as the transition of the technology from research laboratories into clinical settings with bona fide CRISPR-based therapeutics.10,11 These tipping points triggered by specific publications and technology development define distinct time periods that provide useful to assess the dynamic evolution of the field.25,28
An interwoven network of collaborative authors
Next, we carried out a co-authorship network analysis to delve into the collaborative efforts driving contributions by the 48 most prolific and impactful authors over time (Fig. 2 and Table 1). On a global basis, investigating publication patterns across these authors (as defined by number of publications, citations, and h-index within the field), we note extensive and interconnected collaborative networks, with most authors engaged in several collaborative efforts. Actually, it appears the most influential authors collaborate with other key contributing authors in interconnected and overlapping authorship networks (Fig. 2). Interestingly, many “early” authors who were active in the field prior to 2013 originally focused on CRISPR biology and mechanisms of action continue to do so (Fig. 2), whereas distinct collaborative networks that fueled the rise of CRISPR-based genome editing technologies in parallel (Fig. 2A) now directly overlap in topics of interest (Fig. 2B). Noteworthy, the early community-wide focus on Cas9-based genome editing was comprising both overlapping and competitive interests, which created an IP challenge regarding licensing and freedom to operate for the technology,12–14 which presumably prompted searches for novel Cas effectors. Interestingly, while some believe that the CRISPR IP challenges are a scientific hurdle that may have stifled innovation, the data suggest that it may rather have pushed the community toward actively mining for alternatives while not precluding its broad adoption by diverse academic groups across the globe. Those initially established Cas12 as an alternative technology and recently unearthed new CRISPR-Cas types based on Cas13, Cas14, and others,9,29 suggesting a needs-based innovative push rather than a limiting competitive constraint.
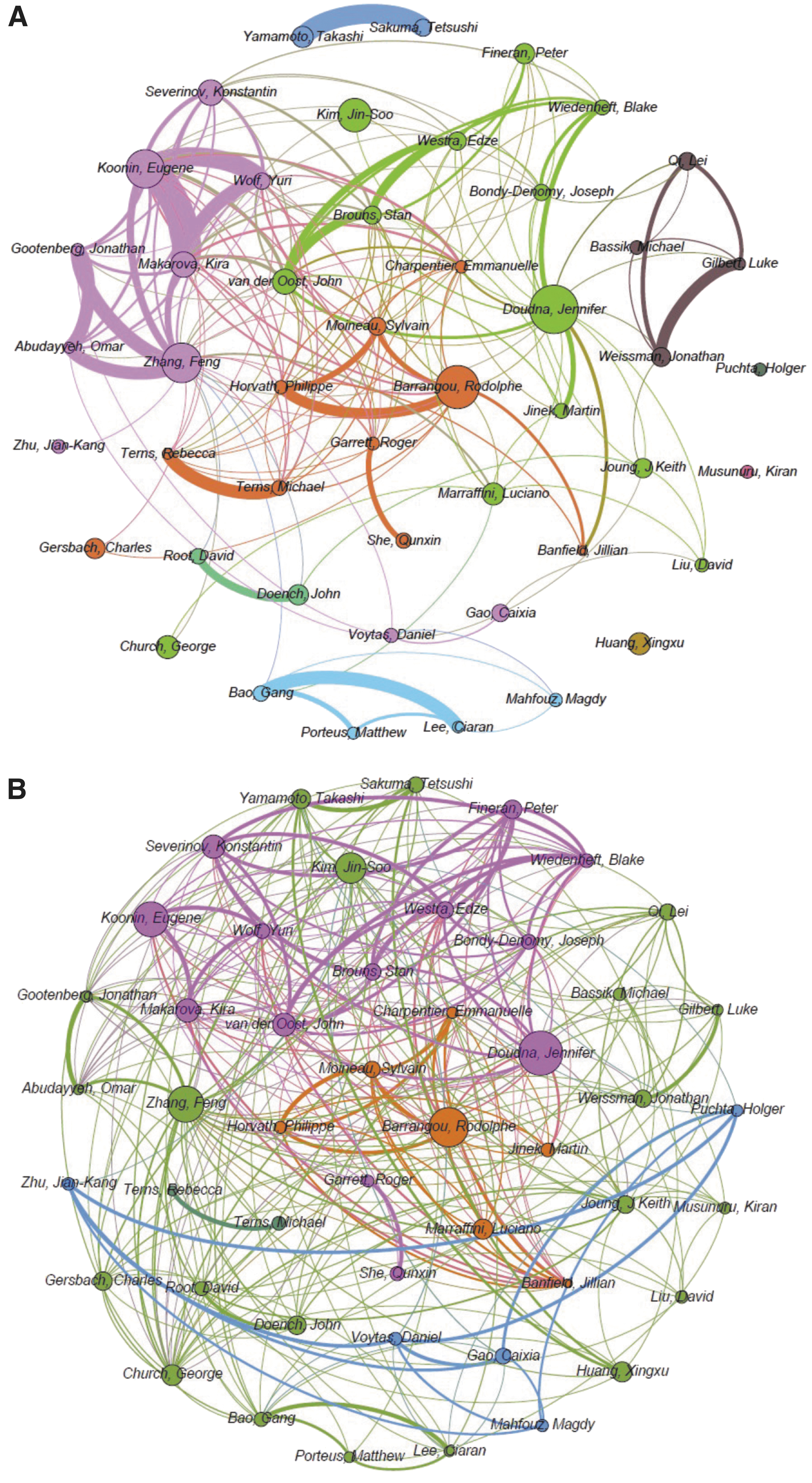
Collaborative authorship networks between the 48 most impactfully prolific CRISPR researchers whose H-index within this topic is more than 20 since 2000.
The number in parentheses indicates the rank in each period.
RE, records; AC, average citations per item; HI, H-index.
Some of the most impactful contributions made by these influential authors can be captured by analyzing the most-cited papers in the field (Table 2) over the three aforementioned eras and the journals in which they have been published (Supplementary Table S3). The early contributions primarily consist of seminal studies establishing CRISPR-Cas as the adaptive immune system in bacteria,7,28 providing DNA-encoded, RNA-mediated, nucleic acid targeting, culminating in 2012 with the development of the sgRNA:Cas9 programmable CRISPR effector. 4 This technology was used in 2013 for genome editing5,6 and shortly thereafter for transcriptional control and high-throughput screens. In the past 2 years, base editing technologies have been on the rise, primarily fueled by the rapid ascent of engineered Cas effectors from the David Liu lab (Tables 1 and 2).9,26,27 Inevitably, the most-cited manuscripts have been research papers published in high-profile journals contributed by prolific authors, together with a few noteworthy reviews and resource-focused papers (Table 2).
Top, 2000–2012; Middle, 2013–2018; Bottom, 2019–2020 (updated March 25, 2021).
Predictably, citation patterns for most highly cited papers in the space reflect the rise of genome editing, notably the rapid explosion in 2013–2014 (Fig. 1). These papers were published in the most influential journals in the world (Supplementary Table S3). Impressively, the most-cited early CRISPR studies were also published in these journals, and they have been and continue to be the most influential journals in this field (Fig. 1 and Supplementary Table S3), despite fundamental shifts in topics of interest and the vast expansion of the contributing authors pool, as well as a diversified and more global readership (Fig. 2). To date, these papers reflect early work, mostly on development of the sgRNA:Cas9 technology, and its use and rapid adoption for genome editing in human cells, with the majority of the most-cited papers published within the first 2 years of the CRISPR craze (Fig. 1B).
In order to delve more into the key organisms, topics, and genes subjected to the most attention in genome editing, we mined the published data and show that human cells are the primary organism of interest for the bulk of genome editing studies, predictably followed by mouse as the canonical proxy animal model for human studies (Supplementary Fig. S1). Noteworthy, studies focused on humans and mice represent 10 times more than all other organisms of interest in CRISPR research, reflecting the heavy focus on human disease and medical applications, notwithstanding interest in and potential for other areas such as agriculture. Actually, this suggests that there is perhaps perplexing under-exploitation or an adoption lag in other areas of interest, such as microbiology, which is ironically where these systems broadly occur and were originally characterized and repurposed. Next, we focused on key diseases of interest in these studies and determined that cancer-related research accounts for the majority of the studies, followed by genetic disease, and infectious disease, including viral infections (Supplementary Fig. S1). This is further corroborated by the top 10 list of genes most associated with genome editing research (Supplementary Fig. S1), notably the most studied trio: TP53 (the most popular tumor suppressor), AKT (protein kinase B), and MYC (proto-oncogene transcription factor).
Emergence of networks of divergent genome editing topics
To gain bibliometric insights into how the field evolved and morphed over time, we used SEP analysis (see Methods) to trace the evolution of topics of scientific interest in these published studies by identifying clusters of linguistic predecessor–descendant topical relationships. 20 This allowed tracking of convergence and divergence of research topics on genome editing and connections among these topics over time (Fig. 3 and Supplementary Fig. S1). This analysis revealed the existence of nine topic communities that have evolved over the three time periods discussed previously. First, the field started with seminal bacterial work that occurred prior to 2012, which focused on adaptive immunity. This community topic is at the core of the network, and initially encompassed foundational topics such as Cas nuclease, acquired immunity, and Escherichia coli (see the pink cluster at the center of Fig. 3 and Supplementary Fig. S1). This core gave rise to the sgRNA:Cas9 genome editing technology, a tipping point for the field, which emerged as a new topic in 2013, centered on guide RNA (derived from the single-guide RNA technology in the context of tracrRNA and crRNA), and links to incumbent genome editing technologies such as ZFNs and TALENs (see the green cluster, Fig. 3). Over time, the core also gave rise to a community focused on screens (genetic screens, high-throughput screens, center right purple cluster). Likewise, the core cluster also gave rise to a community topic focused on transcriptional control, relatively early on with the rise in 2014 of a transcription-focused cluster encompassing gene expression, gene regulation, transcription factors, and transcriptional regulators (center left, blue). Later on, as the technology evolved and matured, application-focused clusters arose, focusing on gene therapies, viral diseases, and neurodegenerative diseases.
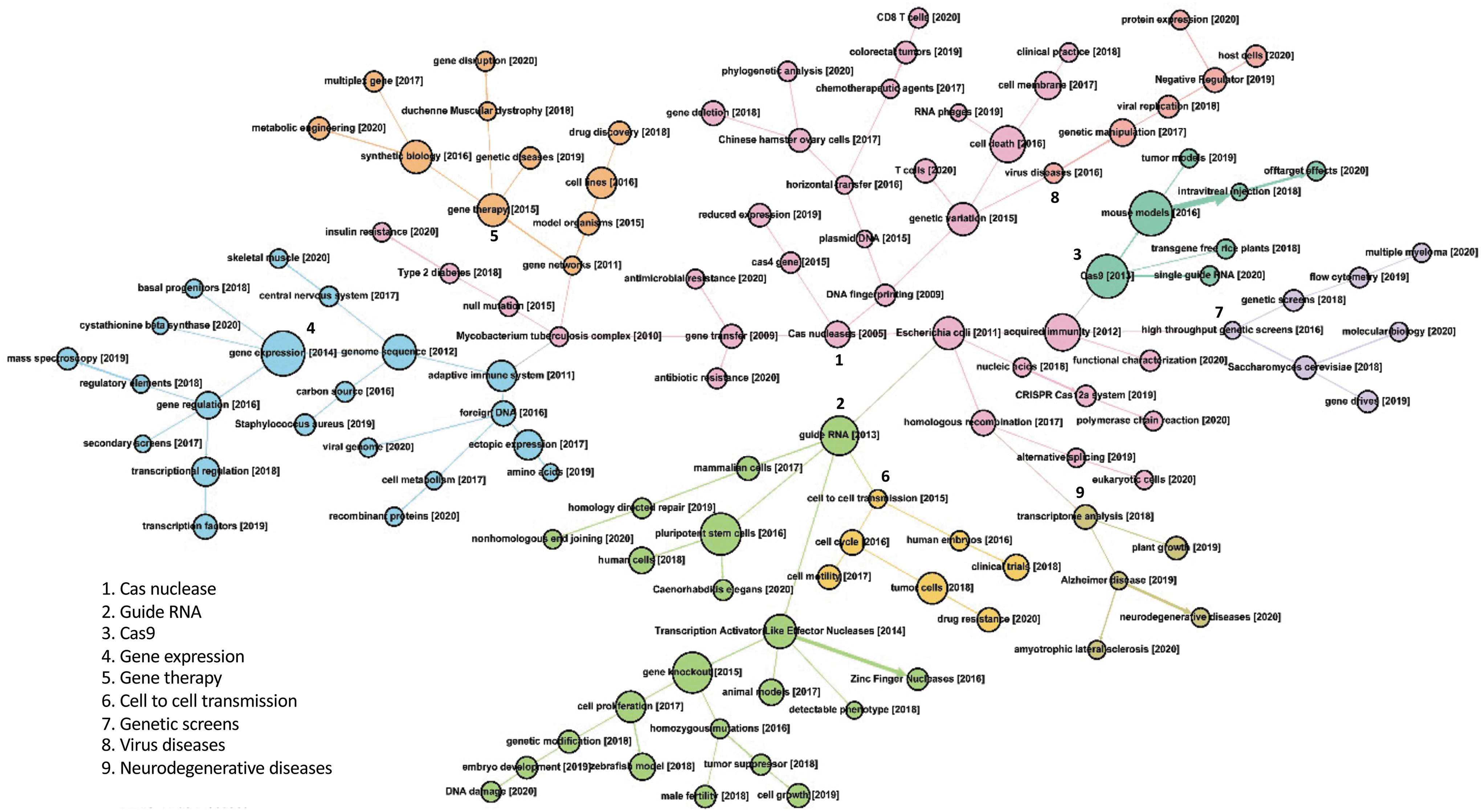
Scientific evolutionary pathway analysis of CRISPR and genome editing topics over time. Nine topic communities are represented using distinct colors, connected over time. Topics are linked using predecessor–descendant relationships defined by the literature patterns.
Analysis of similarity measurements (Supplementary Table S4) between these topic communities reveals how disruptive CRISPR technology is, given the diversity of distinct clusters that arose from the original core cluster, and the relatively low level of similarity observed between and across these 119 topics. This is further supported by the low level of similarity observed between topics across time periods (Supplementary Table S4). The recent increase in topics in the past 2 years (46 new topics in 2 years compared to 64 topics spanning the explosive 2013–2018 period) likely indicates continued disruptive innovation and expansion of this technology into new areas of research, as well as novel and diversified applications. This is consistent with the development of novel technologies (e.g., base editing), the continued dissemination of CRISPR technologies across the globe (e.g., Addgene distribution), and the transition to applications, especially in therapeutic settings with CRISPR-based diagnostics, antivirals, and gene therapies all with clinical ambition in the short term. Critically, it is important to note the cross-referencing of the various visualization modalities and tabular lists of entries throughout our tables and figures that consistently identify the same key factors fueling the genome editing revolution, and robustly establish the seminal studies and technological developments that have shaped this morphing subject over time.
Despite the observed congruence, the SEP algorithm relies on natural language processing techniques that are impacted by writing style and biases, as well as inconsistent use of terminology by different groups of authors, which can lead to synonyms being redundant and accounted for separately. For example, there are entries related to transcription that encompass transcriptional control, gene expression, gene regulation, and transcriptional regulation. There are also several connections between seemingly unrelated topics due to language biases and topic-related complexity inherent to the same technology being used in unrelated organisms. There are also multiple examples of confounding coverage of topics that are often discussed together but are not systematically linked, such as human embryos and clinical trials being discussed together without being codependent. Thus, the complexity of a broadly applicable tool must be deciphered and interpreted by the expert reader to account for otherwise unrelated topics and verbiage. Human interpretation is also important to assess fully the impact and influential contributions of individual authors and select manuscripts in order to account for quantitative shortcomings and biases inherent to citation numbers, indexes, and impact factors. Indeed, qualitative insights should be used by the reader to complement quantitative metrics in the spirit of the Leiden Manifesto. 30 This manifesto highlights the need to rely on expert assessment to overcome bias tendencies and untangle conceptual ambiguity and uncertainty.
In several instances, there are connections that seems counterintuitive and reflect high semantic similarity but not technical dependence or scientific derivation. Indeed, sets of authors can share similar language biases, such as clinically relevant settings for patient sampling in medical applications for the epidemiological study of Mycobacterium tuberculosis and the implementation of genome editing for human gene therapies, linking two seemingly unrelated clusters because the authors share linguistic biases and keywords. Likewise, the link between Cas nucleases and DNA fingerprinting reflects the early use of CRISPR spacer hypervariability for genotyping and not the use of Cas proteins for molecular fingerprinting. This high semantic similarity need not reflect bona fide technical overlap or dependency, and can reveal linguistic biases, or indicate subsequent uses and applications of derived tools and technologies, including their eventual use in diverse model organisms. The latter explains the unexpected appearance of Saccharomyces cerevisiae, Caenorhabditis elegans, zebrafish, Chinese hamster ovary cells, and others throughout topic clusters. Some of the topical lineages shown reflect topical descendance within the CRISPR literature that evolved from a technical basis (using various Cas effectors as tools) to applications of these technologies in model organisms and cells. To a similar extent, select topics of interest to specific groups of authors and readers can be linked through SEP analyses such as human embryos and clinical trials, though they need not be codependent (current clinical trials are not based on CRISPR-edited human embryos). So, both applications and implications can entangle topic connections. In some cases, the appearance of a newly coined term reveals tipping points that created new sets of topics, notably the development of the guide RNA technology and the nomenclature update that reclassified most Cas proteins, including Cas5/Csn1 as Cas9.
While some literature topics have arisen faster than CRISPR, such as the recent COVID19-related literature, 31 the speed of the adoption of the CRISPR technology, as much as the rise of the CRISPR-related literature, is noteworthy. The speed of the work in this field has been invoked as a distinguishing feature, but perhaps the most striking aspect is the adoption and democratization of the technology itself, which is captured by the rise in the number of citations and publications, as well as Addgene shipments.1,2
Discussion
Altogether, these results provide insights into the key factors driving the evolution of CRISPR and illustrate how a diverse community of collaborative scientists is globally adopting this disruptive technology and implementing it in various organisms of interest across applications. This analysis illustrates how bibliometrics can identify key individuals, topics, and papers that dynamically shape a morphing research field and decipher rising trends impacting the historical trajectory of a field and untangle emerging applications.
The data presented here provide strong support that this is a bona fide emerging technology as defined by key attributes. 32 Indeed, all five defining elements of an emerging technology are met, with: (1) radical novelty, near-instant replacement of incumbent editing technologies, with aggressive pursuit of IP and topic diversification; (2) fast growth, as documented by publications, citations, and Addgene distribution patterns; (3) coherence, supported by overlapping collaborative authorship networks, as well as interconnected topics derived from a common core; (4) prominent impact, with enthusiastic commercialization in several industries spanning medicine, agriculture, and biotechnology, as well as global adoption in academia and industry and the momentous 2020 Nobel Prize in Chemistry for two selected CRISPR pioneers; and (5) uncertainty and ambiguity, as documented by IP issues, discussions related to regulatory frameworks for, and societal implications of, the various applications of genome editing. 32 Importantly, the evolution of the topic map over the three aforementioned time periods further endorses the emerging technology attributes of genome editing. Indeed, predecessor topics created during the first time period established a scientific foundation for the field (coherence), with evolution over the next two time periods radically spearheading into various directions (radical novelty), with rapidly increasing number of descendant topics (fast growth), giving rise to diverse research foci.
The eclectic community diversity is noteworthy in terms of institutional affiliations, geographical location, and scientific topics of interest, which collaborations transcend, as illustrated by co-authorship patterns. Yet, the overall primary focus is mostly on human therapeutic applications, reflecting the tremendous potential of genome editing implementation in the clinic, and the need to deploy CRISPR therapies for patients afflicted by genetic diseases. With Food and Drug Administration–enabled trials actively underway, confidence in regulatory agencies and progressing public engagement dialogues encompassing ethical, legal, and societal implications,33,34 we anticipate the literature will continue to expand and hopefully document larger and broad clinical success in the near future, as well as fuel applications in agriculture and sustainability.
Footnotes
Acknowledgment
The authors would like to acknowledge their lab members, collaborators, and colleagues throughout the community for fruitful discussions and insightful opinions.
Author Disclosure Statement
R.B. is a co-founder of Intellia Therapeutics, Locus Biosciences, TreeCo, Ancilia Biosciences, and CRISPR Biotechnologies, and is a shareholder of Caribou Biosciences and Inari Ag. A.P. is a shareholder of Search Technology, Inc. No competing interests exist for the remaining authors.
Funding Information
Y.H. acknowledges support from the National Natural Science Foundation of China (Grant No. 72004169). A.P. acknowledges support from the U.S. National Science Foundation (Award #1759960) to Search Technology, Inc., and Georgia Tech. Y.Z. and M.W. acknowledge the Discovery Early Career Researcher Award granted by the Australian Research Council (Grant No. DE190100994).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.