
Abstract
B lymphocytes are activated and regulated by their interactions with T cells, a process that results in one-way class switching of immunoglobulins (ig) from IgM to IgG, IgE, or IgA. In this study, we show the application of clustered regularly interspaced short palindromic repeat-Cas9-induced nonhomologous end joining in B cells to achieve reverse-directional Ig class switching. By electroporating Cas9 and guide RNA and a Cμ encoding donor into cells, we engineered IgG-secreting human B cell lines to switch to express IgM antibody. This approach offers a new potential path for the production of IgM antibodies.
Introduction
Immunoglobulin (Ig) class switching is the process by which a B cell changes its antibody expression from IgM to IgA, IgG, or IgE. The mechanism underlying this phenomenon, the so-called class switch recombination (CSR), allows the immunoglobulin heavy chain (IgH) constant region exon of an antibody to change but preserves the antigen specificity.1,2 The constant region exon for IgM (Cμ) is located upstream of the exons of IgD, IgG, IgE, and IgA isotypes, and CSR occurs between the switch or S regions.
Breaks and rejoining the DNA of the two S regions lead to a rearranged constant heavy chain locus. The DNA segment between the two switch regions is irreversibly deleted, indicating that Ig class switching is unidirectional (e.g., from IgM to IgG). During humoral immune responses, antigen-specific IgM is first produced, followed by the production of other Ig isotypes. Under physiological conditions, IgG-producing B cells cannot switch to express IgM antibody.
The bacterial type II clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 (CRISPR-associated protein 9) system is a versatile tool for genome editing in human cells.3,4 CRISPR-Cas9-induced double-stranded breaks can be repaired by homology-directed repair (HDR) or nonhomologous end joining (NHEJ). HDR-mediated gene replacement and NHEJ-induced knockout are most common in CRISPR-Cas9 applications.5,6
High-efficiency knock-in of exogenous DNA sequences into a selected genomic locus has been recently achieved by employing CRISPR-Cas9-induced NHEJ.7–11 In this method, a donor plasmid and targeted genomic loci harboring the same guide RNA (gRNA) recognition sequence are simultaneously cut by Cas9 and then rejoined through NHEJ, resulting in targeted integration of defined DNA sequences.
Genome editing has remarkably expanded our ability to manipulate DNA and has shown great potential in the field of B cell engineering.12–18 Cheong et al demonstrated that IgM+ B cells can be engineered to switch to express IgG and IgA using CRISPR-Cas9-induced CSR of the IgH chain. 19 However, whether B cells can switch from IgG to IgM remains unknown. We hypothesized that B cells can be reprogrammed to undergo a reverse directional class switch by inserting a constant region for Ig into the appropriate gene loci. In this study, we show that human B cell lines can be engineered to switch from producing IgG to IgM by CRISPR-Cas9–mediated NHEJ.
Materials and Methods
B cell engineering reagents
A gRNA (5′-ATATTCCACCCAGGTAGTGG-3′) targeting upstream of the Sμ site within the IGH locus of the human genome was selected and designated as Sμ 5′ gRNA, as was used in a previous study. 19 A single guide RNA (sgRNA) was produced using EnGen sgRNA Synthesis Kit (NEB) and Monarch RNA Cleanup Kit (NEB) according to the manufacturer's instructions. The cleavage efficiency of sgRNA was validated by cutting the target sequence with sgRNA and Cas9 in vitro.
In brief, a PCR amplicon (primers are listed in Supplementary Table S1) spanning the target site was synthesized from genomic DNA using GoTaq Green Master Mix (Promega). The PCR cycling conditions were 95°C for 2 min, followed by 35 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The PCR product was purified with a zymoclean Gel DNA Recovery Kit (Zymo), and then combined with the sgRNA and recombinant Cas9 (NEB), following the manufacturer's instructions. The cleavage efficiency was evaluated by agarose gel electrophoresis.
Donor plasmid
The Cμ insertion cassette consisted of 114 bp upstream of Cμ exons, the full-length coding region of human Cμ (hg38 chr14:105,851,966–105,856,218), a porcine teschovirus-1 2A-green fluorescent protein (P2A-GFP) coding sequence, and 261 bp downstream of Cμ. The Cμ-inserting cassette with two gRNA target sites flanking the 5′ and 3′ sides was synthesized (GenScript) and constructed into pUC57-mini (GenScript) as a donor. The synthesized sequence is listed in Supplementary Table S2. The donor plasmid was validated using sequencing.
Cell culture and electroporation
IgG-producing human B cell lines ARH-77 (CRL-1621) and IM-9 (CCL-159) were purchased from the American Type Culture Collection (Rockville, MD, USA). The ARH-77 cell line was isolated from a patient with IgG plasma cell leukemia. The IM-9 cell line is an Epstein–Barr virus-transformed B lymphoblastoid cell line established from a patient with multiple myeloma. Both cell lines were grown in RPMI 1640 medium (Hyclone) supplemented with 15% fetal bovine serum (Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C and 5% CO2.
For transfection, 2,000,000 cells were suspended in 20 μL electroporation buffer (Celetrix). Then 30 pmol sgRNA was premixed with 60 pmol Cas9-NLS (NEB) at room temperature for 10 min, and then the formed ribonucleoprotein (RNP) and 1 pmol donor plasmid were mixed with the cells and transferred to a 20 μL electroporation tube (Celetrix). Cells were electroporated using an electroporation machine (CTX-1500A LE; Celetrix) at a voltage and pulse time of 440 V and 30 ms, respectively. The cells were immediately seeded into 24-well plates in a warm medium after electroporation.
PCR verification of Cμ inserting and Sanger sequencing
Five days after electroporation, genomic DNA was isolated from transfected cells using the DNeasy Kit (Qiagen) following the manufacturer's instructions. Targeted integration of the Cμ donor at Sμ 5′ (Fig. 1) was assayed through PCR amplification (PCR primers are listed in Supplementary Table S1) using GoTaq Green Master Mix (Promega). In brief, the PCR conditions were 95°C for 2 min, followed by 40 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min.
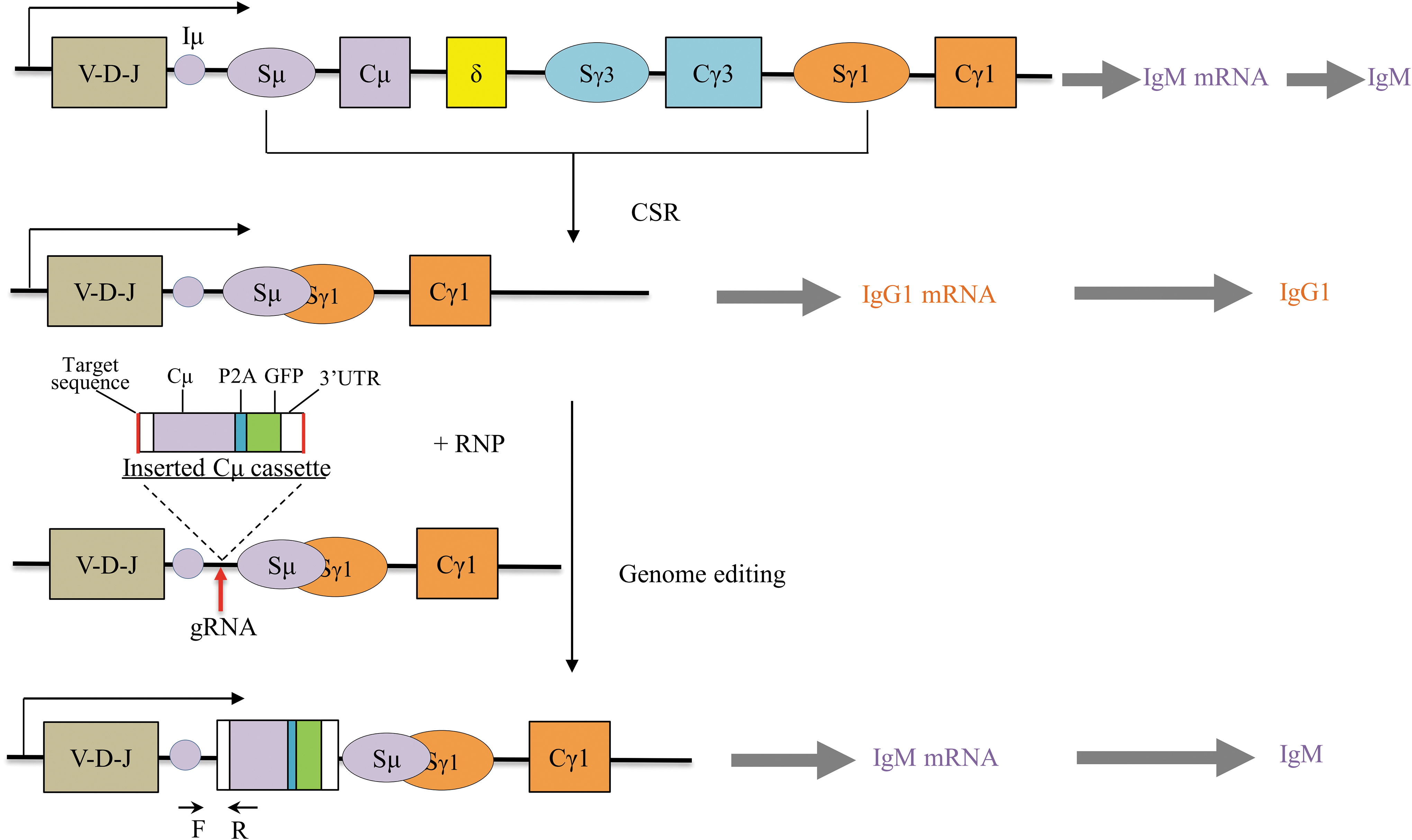
Schematics of CRISPR-Cas9–mediated re-expression of IgM antibody in IgG-producing cells. The red arrow refers to gRNA cleavage site. Black arrows at the bottom indicate the PCR primers designed to validate correct integration. CRISPR, clustered regularly interspaced short palindromic repeat; CSR, class switch recombination; gRNA, guide RNA; RNP, ribonucleoprotein.
For Sanger sequencing, 1 μL of the first PCR product was reamplified with nested PCR primers (listed in Supplementary Table S1) using GoTaq Green Master Mix (Promega) under the following cycling conditions: 95°C for 2 min, followed by 95°C for 30 s, 57°C for 30 s, and 72°C for 20 s, for 30 cycles, and a final extension at 72°C for 5 min. The nested PCR product was recovered using the Zymoclean Gel DNA Recovery Kit (Zymo), cloned into pMD18-T Vector (TAKARA), and five individual bacterial colonies were picked for Sanger sequencing.
Enzyme-linked immunosorbent assay
Fractions of cell culture supernatants were collected 3 and 5 days after transfection. The secreted IgM antibody in the supernatant was detected using the Human IgM ELISA Quantitation Set (Bethyl Laboratories) according to the manufacturer's instructions. The IgM antibody concentration in each sample was determined referencing the standard curve.
Reverse transcription-PCR
RNA was extracted from transfected cells 3 days later using the SV Total RNA Isolation System (Promega) according to the manufacturer's instructions. The extracted RNA was eluted in 100 μL of nuclease-free water and 1 μg was further treated with DNase I (Thermo Fisher Scientific) for 15 min. Finally, 2 μL of the digested RNA was subjected to reverse transcription-PCR (RT-PCR) using the OneStep RT-PCR Kit (Qiagen) following the manufacturer's instructions. Human IgM mRNA and β-2 microglobulin mRNA were amplified using specific primers (listed in Supplementary Table S1).
In brief, the reaction mixture contained 2 μL of RNA template, 28 μL of RNase-free water, 10 μL of 5 × buffer, 2 μL of dNTP mix, 2 μL of enzyme mix, and 3 μL of each primer. The cycling conditions were as follows: 50°C for 30 min, 95°C for 15 min, followed by 40 cycles of 94°C for 30 s, 61°C (IgM mRNA)/54°C (β-2 microglobulin mRNA) for 30 s, and 72°C for 60 s, with a final extension at 72°C for 10 min.
Fluorescence-activated cell sorting
Flow cytometry was carried out 96 h after electrotransformation. To enrich the living cells, cells were stained with 7-aminoactinomycin D (7-AAD; BioLegend) for 5 min on ice, and subjected to fluorescence-activated cell sorting (FACS) using LE-MA900FP (SONY). The cells were gated using 7-AAD and GFP. The 7-AAD− and GFP+ cells were single sorted and further cultured in RPMI 1640 containing 20% fetal bovine serum. Data were analyzed using FlowJo software (FlowJo).
Immunofluorescence microscopy
Cells were collected 120 h post-transfection and washed three times with phosphate-buffered saline (PBS), then fixed with 100% methanol at 4°C for 15 min. Cells were washed once with PBS, then twice with 2.5% skim milk powder in PBS. Cells were costained with goat antihuman IgM FITC (Elabscience) polyclonal antibody (1:50) and goat antihuman IgG APC (Jackson ImmunoResearch) polyclonal antibody (1:50) for 1 h at room temperature. After washing three times with PBS, cells were resuspended in 100 μL PBS and mounted on a glass slide using mounting medium containing DAPI (Vector Laboratories). Images were captured with a high-resolution CCD camera mounted on a fluorescent microscope (Zeiss Axio Scope A1).
Results
Targeting strategy and donor plasmid design
IgG-secreting cell lines ARH-77 and IM-9 retained the Ig joining (J)—Sμ region, which contains the IgH enhancer μ (Eμ) and initiation μ exon (Iμ). Theoretically, inserting the Cμ coding sequence into the region between Iμ and Sμ would allow the retranscription and reexpression of Cμ (Fig. 1). The Cμ insertion cassette contained a splice junction at the 5′ end to aid splicing to the upstream heavy chain J region (Fig. 1). A 261 bp 3′ UTR fragment was also included to enable immunoglobulin μ-chain expression.
We also fused P2A-GFP to Cμ to allow monitoring of the expression of IgM or selection of engineered cells. NHEJ-mediated insertion relies on the simultaneous cleavage of the donor plasmid and the chromosome target site, followed by ligation of the same DNA ends. Thus, the Cμ cassette was additionally flanked by two gRNA-targeting sequences to induce double cut of the donor plasmid.
Integration of the Cμ cassette in IgG-producing B cell lines
The cleavage efficiency of Sμ 5′ gRNA was high, as shown in Supplementary Figure S1. We transfected ARH-77 and IM-9 cell lines with Sμ 5′ gRNA precomplexed with Cas9-NLS and a donor plasmid containing the Cμ cassette. The expected 5′ junctions formed by the IgHM and Cμ inserting cassettes were detected by PCR amplification in the presence of both sgRNA/Cas9 and donor plasmid (Fig. 2A). The expected amplicon length was 564 bp.

Validation of Cμ cassette integration in the transfected cells by PCR and sequencing. Total genomic DNA was isolated 5 days later from cells mock treated or electroporated with RNP and donor plasmid. A 5′ integration PCR was carried out.
Nonspecific bands were also observed. We further conducted a nested PCR, which produced a specific band with a theoretical size of 136 bp (Fig. 2B). The target fragments were purified, cloned into the pMD18-T vector, and sequenced. Clones with precise integration containing the expected target chromosome sequence and donor plasmid sequence were observed (Fig. 2C), confirming the successful integration of the Cμ cassette into the target genomic locus of IgG-producing B cell lines.
CRISPR-Cas9–mediated IgM expression in IgG-producing human B cell lines
To investigate whether IgG-secreted B cells can be converted to express IgM antibodies, we engineered the IGH locus by inserting a Cμ-expressing cassette through the CRISPR-Cas9 system. As expected, human IgM antibodies were detected by ELISA in culture supernatant from cells electroporated with Cas9 and sgRNA targeting the Iμ–Sμ region and donor plasmid 3 days later (Fig. 3A). For each transfected cell line, the concentration of IgM antibody was increased through additional culture. We also confirmed the expression of human IgM mRNA by RT-PCR in the engineered cell lines. 20
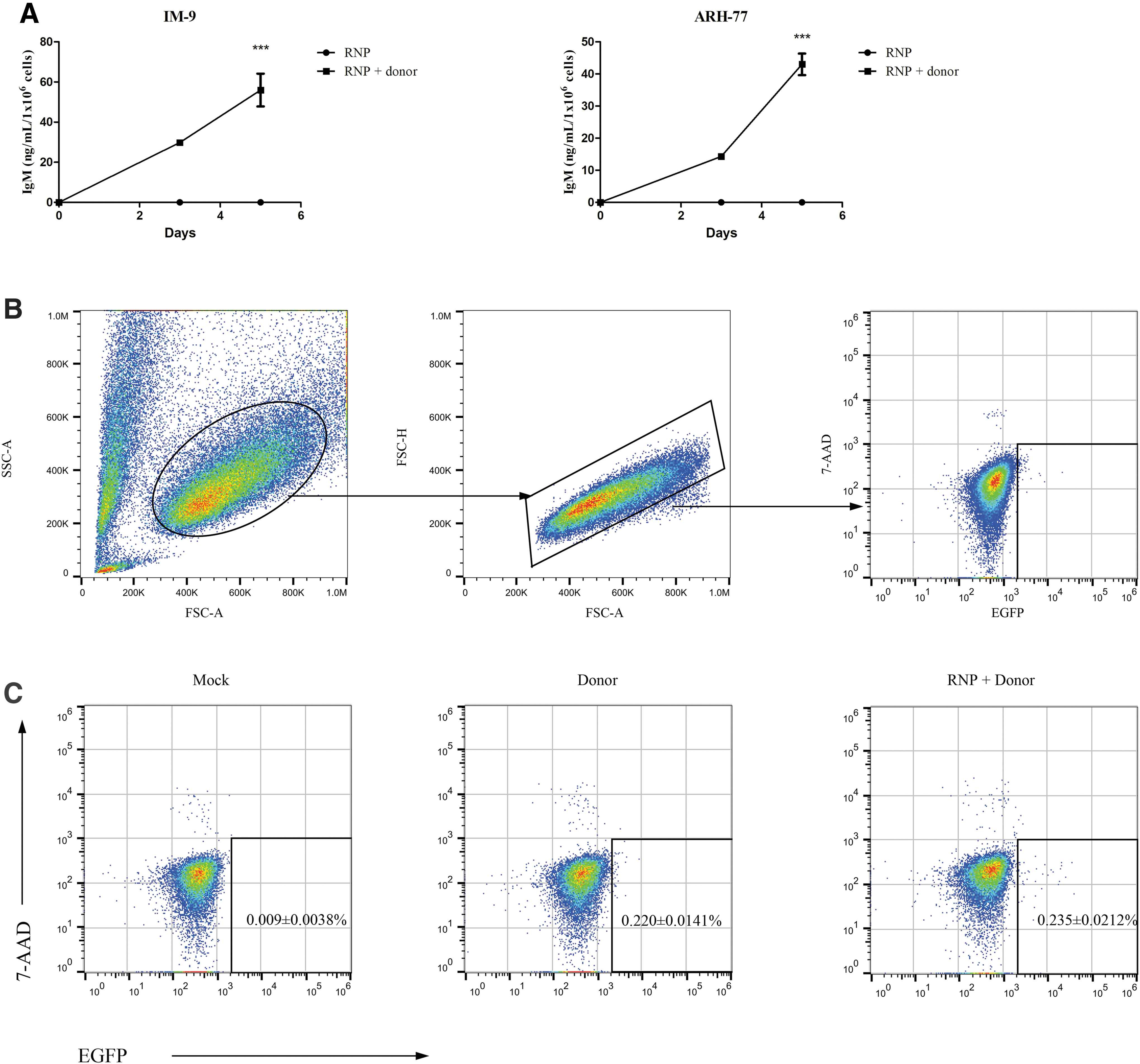
CRISPR-Cas9–mediated NHEJ induces IgM production in IgG-secreting cells.
If a DNA segment of Cμ was extracted along with RNA, the RT-PCR would yield another product with 365 bp (amplified from DNA template) in addition to the expected amplicon of 186 bp (generated from the mRNA template). To exclude the effect of the transfected Cμ cassette on IgM mRNA detection, the purified RNA was further digested with DNAse I and subjected to RT-PCR. As shown in Figure 4, a specific band with the expected size of 186 bp was detected, indicating the presence of IgM mRNA. Residual undigested DNA may explain the weak band at 365 bp.
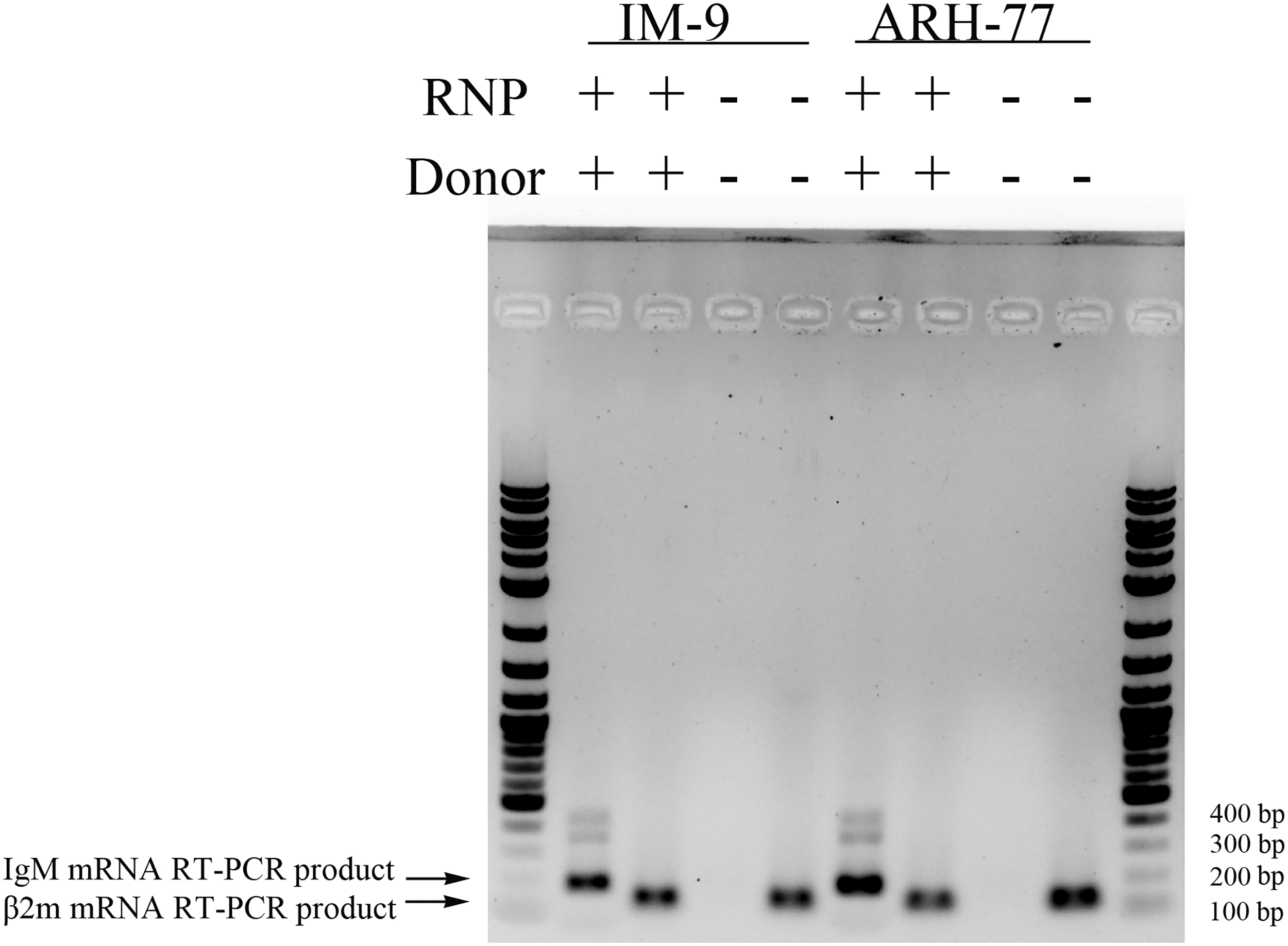
CRISPR-Cas9-induced NHEJ enables human IgM mRNA expression in IgG-producing cells. Total RNA was extracted 3 days later from cells transfected with RNP and donor plasmid or mock treated. Human IgM mRNA and β-2 microglobulin were detected by RT-PCR using specific primers. The weak band between 400 and 500 bp is a nonspecific PCR product. β2m, β-2 microglobulin; RT-PCR, reverse transcription-PCR.
Next, we attempted to enrich and isolate single engineered IM-9 cells using FACS. Ninety-six hours after electroporation, cells were collected and subjected to flow cytometry. The results are shown in Figure 3. Compared with mock controls, a very small fraction of living cells expressing GFP were observed both in the RNP and donor-treated group and the donor-only controls (Fig. 3B, C). And their frequencies of positive cells were comparable (0.235% ± 0.0212% vs. 0.220% ± 0.0141%). However, it is notable that the absolute number of positive cells with strong GFP signals (104–105) from the RNP and donor-treated group was larger than that from the donor-only transfected controls.
The flow cytometry result (Fig. 3C) might be explained by two reasons: (1) the knock-in frequency of Cμ cassette was rather low in this lymphoid cell line and (2) the fused P2A-GFP was not successfully processed and expressed in the Cμ cassette inserted B cells. We collected and sorted 96 GFP+7-AAD− cells; however, most of them died after sorting, and 2 cells developed into colonies of ∼8–30 cells but did not grow any further. Technically, lymphoblastoid cell lines are difficult to subclone from single cells. Moreover, electrotransfection and FACS are harmful to cells. Feeder cells may be used to support cell growth.
A direct stain for the expression of IgM by using an anti-IgM antibody in the gene-edited cells was further carried out. Compared with untreated control, a small percentage of cells (2.12%–2.25%) treated with RNP and donor expressing human IgM (Green) were observed (Fig. 5).
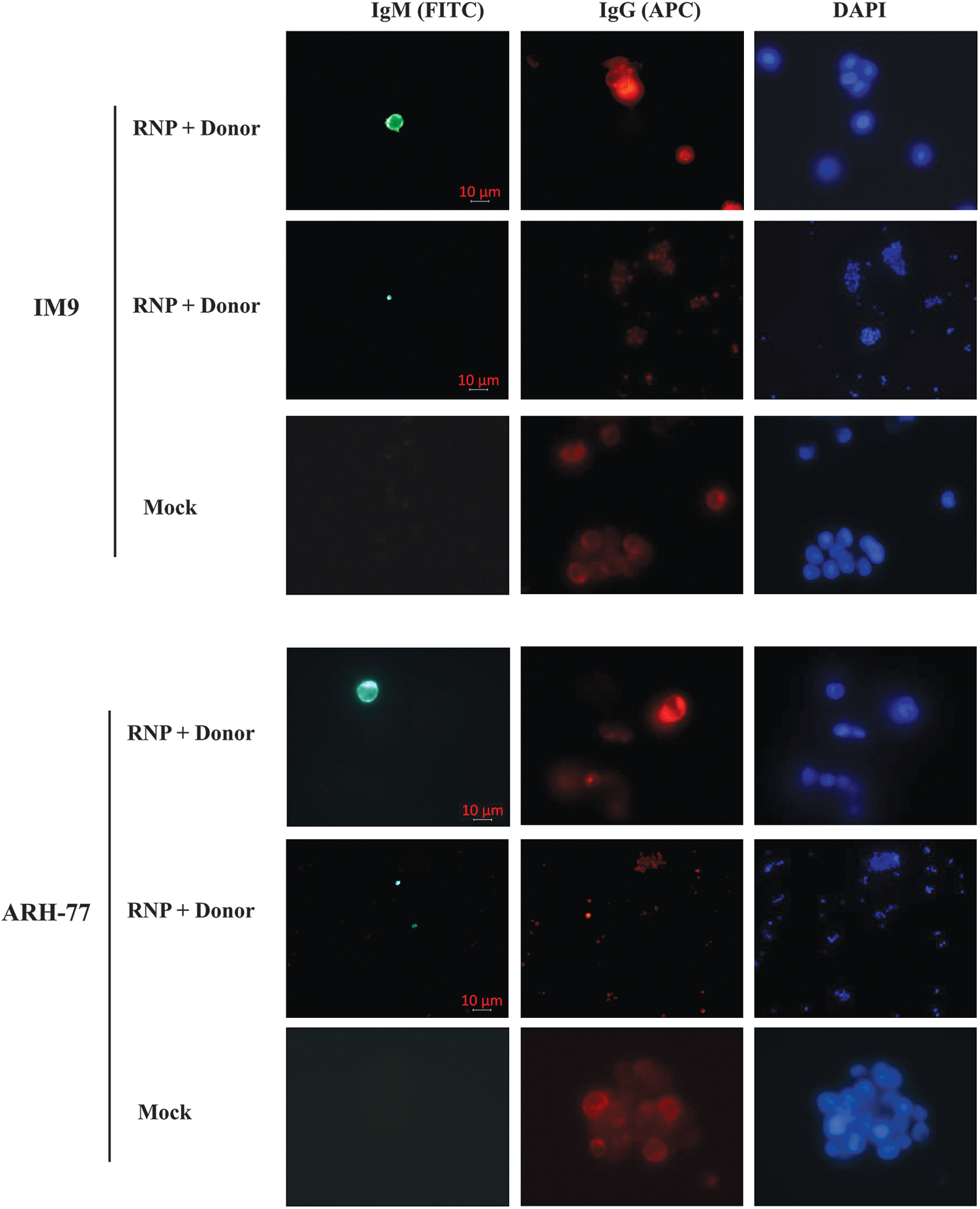
Immunofluorescence microscopy of IgM expression in genome-engineered human B cells. Five days after transfection, B cells were coincubated with anti-IgM FITC and anti-IgG APC polyclonal antibodies. Green fluorescence (FITC) refers to the IgM antibody, red (APC) indicates the IgG antibody, and blue denotes the nucleus (DAPI). APC, allophycocyanin; DAPI, 4′,6-diamidino-2-phenylindole; FITC, fluorescein isothiocyanate.
In summary, the results from human B cell lines demonstrated that correct integration of Cμ can be achieved by the CRISPR-Cas9 system, and that IgG-producing B cells can be engineered to switch to express IgM antibodies.
Discussion
Currently, human monoclonal antibodies are preferentially generated by single-cell B cell receptor cloning or by the immortalization of memory B cells.21–24 CRISPR-Cas9 enables insertion of an Ig expression cassette at the Ig locus or other gene loci in B cells, resulting in defined monoclonal antibody expression.13–15,17,18 In this study, we demonstrated that IgG-secreting human B cell lines could be engineered to become IgM-producing cells by integration of a full Cμ coding sequence using CRISPR-Cas9–coupled NHEJ repair. Our data provide another potential path for IgM antibody production in biomedical research and clinical applications. For example, pathogen-specific IgM assays are important for the diagnosis of acute infections.
However, these tests are commonly hampered by the limited availability of pathogen-reactive human IgM-positive control specimens, because these polyclonal antibodies are only present during acute infection and are difficult to obtain. Monoclonal antibodies produced using the single-cell B cell receptor cloning method are not suitable for control purposes. This is because they are only recognized by specific antigens and may not be commutable for different antibody detection kits. Pathogen-specific polyclonal IgG and/or IgM antibodies can be generated by immortalized memory B cells and/or plasma cells.21,22,25
By applying the proposed method, it is possible to switch these IgG-producing memory B cells to produce antigen-specific polyclonal IgM antibodies as control materials when plasma cells are not available. IgM antibodies also have therapeutic potential. Because of their greater avidity against antigens composed of polymeric or repeated motifs over IgG, ∼20 IgM antibodies have advanced to clinical trials for the therapy of oncology, infectious disease, and autoimmune disease. 26
Lymphoid cell lines are difficult to transfect using the cationic–liposome method; thus, we delivered engineering materials into human B cell lines using electroporation,27,28 a rather simple method. The frequency of successfully genome engineered cells was not high as indicated by the immunofluorescence results (Fig. 5), which could be explained by the transient but harmful transfection method used, and the rather large insert (5431 bp) knock-in.
A previous study demonstrated that the NHEJ-based knock-in frequencies decreased as the size of the integration DNA increased. 8 CRISPR-Cas9 delivery and knock-in frequencies may be improved by using retroviruses or lentiviruses, although they are much more complicated. We did not get single pure IgM-producing clones; however, the direct detection of IgM antibody and IgM mRNA supports the expression of IgM antibody in the genome-edited B cells.
As a proof-of-concept study, we demonstrated that IgG-secreting human B cells can be engineered to undergo reverse directional CSR using CRISPR-Cas9 technology.
Footnotes
Authors' Contributions
Study conception and design were carried out by G.L. and J.L.; experiment conduction was done by G.L., Y.H., R.P., J.Z., and D.L.; analysis and interpretation of results were taken care by G.L., K.Z., and J.L.; draft article preparation was carried out by G.L. and K.Z.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by Beijing Municipal Natural Science Foundation Grant 7192188 (G.L.), Beijing Dongcheng District Outstanding Talent Nurturing Program 2019WJGW-10-01 (G.L.), National Natural Science Foundation of China Grant 81974319 (J.L.), National Nature Science Foundation of China Grant 81902145 (R.P.), and Beijing Municipal Nature Science Foundation Grant 7204299 (R.P.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.