
Abstract
The design of CRISPR-Cas9 guide RNAs is not trivial and is a computationally demanding task. Design tools need to identify target sequences that will maximize the likelihood of obtaining the desired cut, while minimizing off-target risk. There is a need for a tool that can meet both objectives while remaining practical to use on large genomes. In this study, we present Crackling, a new method that is more suitable for meeting these objectives. We test its performance on 12 genomes and on data from validation studies. Crackling maximizes guide efficiency by combining multiple scoring approaches. On experimental data, the guides it selects are better than those selected by others. It also incorporates Inverted Signature Slice Lists (ISSL) for faster off-target scoring. ISSL provides a gain of an order of magnitude in speed compared with other popular tools, such as Cas-OFFinder, Crisflash, and FlashFry, while preserving the same level of accuracy. Overall, this makes Crackling a faster and better method to design guide RNAs at scale. Crackling is available at https://github.com/bmds-lab/Crackling under the Berkeley Software Distribution (BSD) 3-Clause license.
Introduction
The design of guide RNA is a crucial step for any CRISPR experiment. However, guide design is not trivial, as the efficiency and specificity of guides are crucial properties. Efficiency broadly refers to the guide correctly binding to the targeted region and to the endonuclease. This is influenced by a number of factors that depend both on the guide itself (e.g., secondary structure) and on the targeted region (e.g., chromatin accessibility). Specificity refers to the ability of the guide RNA to have minimal off-target modifications. This means not only that the targeted sequence must be unique but also that closely related sequences (typically less than four mismatches) elsewhere in the genome must be carefully considered. There exist many computational pipelines to assess the properties of CRISPR guides. However, they are sometimes inaccurate and often slow. 1 In this study, we present Crackling, a pipeline designed to rapidly analyze large-sized genomes, without sacrificing accuracy.
We recently benchmarked existing guide design tools.
1
Our two main findings were as follows:
When considering efficiency, for any given genomic sequence, there is a limited overlap between the set of guides that each tool produces. Several tools have inadequate filtering on specificity, and when that is considered carefully, it tends to be a computationally expensive task.
The limited overlap between the tools can be exploited for guide selection: when a guide is recommended by multiple tools, there is a higher chance that it will actually be efficient. We explored consensus approaches in detail and found that they provide better guides. 2 However, it is not practical to run several different tools. A recent review suggested the use of a tool that would offer more than one scoring algorithm, and while these tools exist, none of them have explored the possibility of combining scores to achieve an overall gain in precision. 3 Our Crackling method directly integrates multiple scoring approaches and combines their result for improved precision when predicting guide efficiency.
While guide efficiency is critical, the success of a CRISPR experiment is also determined by the specificity of a guide RNA. Several methods have been implemented to tackle evaluating specificity. One of the earliest methods, published in 2014, is Cas-OFFinder, 4 which provides a count of the number of mismatches between each candidate guide and off-target site if the count is less than a predetermined threshold (e.g., four). The results can then be used to get a sense of the off-target risk for each candidate guide.
While identifying off-target sites that are within a fixed number of mismatches is useful, it is not necessarily enough. It has been experimentally shown that the position of the mismatches also matters. 5 A score for this exists; sometimes referred to as the Zhang score, from the corresponding author of that article. To calculate the score, a candidate guide is compared against all potential CRISPR sites in the genome of interest. When a site is at most four mismatches away, a local score is calculated. The sum of local scores provides a measure for the off-target risk of some candidate guide. Methods such as Cas-OFFinder, 4 CRISPRseek 6 and CRISPOR,7,8 and mm10db 9 each use a custom implementation of this score.
In mm10db, the multithreaded function implemented for calculating the score, called findMismatches, terminates early as soon as the running sum of local scores guarantees that the global score will drop below some fixed threshold.
The initial implementation of findMismatches relies on string comparisons. For this article, we have reimplemented it using a binary encoding of the sequences, so that mismatches can be identified more efficiently.
An alternative to the Zhang score is the Cutting Frequency Determination (CFD) score, which accounts for position- and identity-specific mismatches between the guide and off-target sequences. 10 Doench et al. assessed the ability of the score to predict the off-target activity for an independent set of 89 guide sequences. Compared with other popular scores, including the Zhang score, CFD continually performed better when measured using the area under the receiver operating characteristic curve (AUROC), as shown by Doench et al. 10
The Zhang and CFD scores have been used in an array of ensemble learning algorithms, including using simple consensus approaches, and the machine learning algorithms AdaBoost, Random Forest, Support Vector Machine, and Decision Tree. 11 The CFD score was found to be a driver in predictive performance. The simple pairwise combination of the Zhang and CFD scores performed best compared with other techniques and any score taken individually.
Similarly to the Zhang score, instead of calculating an exact global score, it is possible to keep track of the running sum of local CFD scores and terminate early as soon as a guide is guaranteed to end up being rejected.
The recently published Crisflash relies on the Zhang score to evaluate guides, but without the early termination. 12 The authors report that Crisflash can outperform Cas-OFFinder by an order of magnitude on the human genome when more than a couple of 100 guides are supplied. We benchmark the performance of these tools in this study.
Crisflash also provides support for variants, which are not used here. It is also noted that Crisflash relies on a volume of memory that would not be available on a modern workstation (90 Gb), but rather a server cluster.
FlashFry presents a discovery approach that utilizes a table of bit-encoded off-target prefixes. 13 Similar to other methods, a candidate guide is only scored on a given off-target if the number of mismatches is fewer than the specified threshold. The authors claim that FlashFry is able to run two to three orders of magnitude faster than Cas-OFFinder. FlashFry does not report the Zhang score but uses the CFD score.
Other tools such as CRISPRseek, CasOT, and CRISPRitz exist.6,14,15 However, we do not consider them here as published results show that they perform poorly compared with others, which we have included. 12
Materials and Methods
In this article, we present Crackling, a method aimed at addressing the two challenges of efficiency and specificity when designing CRISPR-Cas9 guide RNAs. To increase the efficiency of the set of guides we produce, we have combined three scoring strategies, and to speed up the off-target scoring, we have introduced a solution for constant-time lookup of sequence neighborhoods.
The tool is implemented in Python (version 3) for most steps, with the high-performance off-target scoring in C++. It can run on any platform with minimal dependencies: Bowtie2 for guide realignment onto the input genome and RNAfold for secondary structure prediction.16,17 Where possible, components of the tool are parallelized for improved performance. Preprocessing is minimal, only requiring the input genome to be indexed by Bowtie2 and off-target sites to be indexed for Inverted Signature Slice Lists (ISSL). 18 The ISSL index is simple to build and can be reused at any time for the genome it is constructed for.
Crackling is configured via an initialization file, which we have provided thorough documentation for. Parameters include:
Providing a name to prefix output file names.
An optimization level that dictates how restrictive Crackling is to be when calculating results. A low optimization instructs Crackling to calculate results for all tests, whereas a high optimization progressively filters out tests, on an individual guide basis. The optimization level will influence the run-time.
The consensus approach: selecting which rules, derived from other tools, should be included.
Paths to input and output files.
Program binaries, threading, and batch sizes to avoid saturating memory.
Once configured, the pipeline is called via any command line terminal using the Python v3.7 interpreter. Postprocessing, which provides the ability to annotate guides with any genomic features and to count the number of targeted gene transcripts, is an optional final step, for which we also provide code.
Multiapproach efficiency evaluation
We previously showed that when different tools actively filter or score guides based on their predicted efficiency, they only rarely agree with each other. 1 We also showed that this can be leveraged for consensus-based approaches that combine the output of multiple tools. 2 However, having to install and run multiple tools may not always be practical.
Based on the results of this consensus study and the need for computational performance, Crackling directly incorporates three scoring approaches and only recommends candidate guides that are accepted by at least two of the approaches:
As in CHOPCHOP, we also look for the presence of a guanine at position 20. 19
We select for positively scored guides based on the model from sgRNAScorer 2.0. 20
We use the filtering steps from mm10db (GC content, secondary structure, etc.), and only accept candidate guides that pass all steps. 9
Other consensus-based approaches can be easily incorporated by modifying the default configuration. Table 5 describes the precision (the proportion of selected guides that are truly efficient), recall (the proportion of efficient guides that are selected), and F1 score (the harmonic mean of precision and recall) for each configuration.
For each configuration, we explored the number of guides that Crackling reported per gene (for the Oryza sativa genome), before considering off-target effects. For the strictest configuration, where all three scoring approaches must agree, Crackling designed guides for 93.9% of genes. This configuration also reported at least three guides for 90% of genes, which allows for a triple-target strategy delivering almost perfect knockout efficiency. 9 The default configuration designed guides for 96.4% of the genes. Figure 1 describes the number of guides per gene for all configurations.

Number of guides per gene, for each consensus configuration, shown as proportions across the Oryza sativa genome. Range boundaries are inclusive.
The performance of Crackling may change depending on the configuration chosen. See Supplementary Figure S1 for the run-time of Crackling when ran in each of the configurations.
The sgRNAScorer 2.0 model is included in the Crackling code repository, in addition to the raw data and a script to retrain the model for version compatibility reasons.
Assessing off-target risk using the Zhang and CFD scores
The Zhang score assesses the off-target risk by considering position-specific mismatches between the candidate guide and CRISPR sites that differ by no more than four mismatches. For each CRISPR site meeting this criterion, a local score is calculated using Equation (1).

where W is an array of position-specific weights, d is the pairwise distance between mismatches, n is the number of mismatches between target and sequence, and M is the list of mismatch positions.
The sum of local scores provides a measure for the off-target risk of some candidate guide. If we denote Q as the set of all off-target sites at most four mismatches away from the candidate sgRNA, then Equation (2) combines all the
The objective is to reject all guides for which the global score is below a fixed threshold
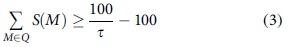
The CFD score is stricter than the Zhang score, as in addition to the position of the mismatch, it also considers the type of mismatch. A global CFD score can be calculated in the same way as the Zhang score—first by calculating local scores for CRISPR sites that are no more than four mismatches away from the candidate guide, as in Equation (4).
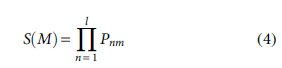
where P is a matrix of percent-active values for positions n to the length of the guide sequence l and mismatch type m. Greater values of
By again using Equation (2) to combine the local scores, a global CFD score can be calculated for the guide. When the score is below a fixed threshold
Multiapproach specificity evaluation
A plethora of tools have been proposed for predicting off-target activities of CRISPR-Cas9. Each has been critically evaluated using data that include measured editing outcomes. Zhang et al. assessed an array of techniques for combining the tools, including using simple consensus approaches, and machine learning algorithms. 11 The simple pairwise combination of the Zhang and CFD scores was found to be better than other techniques and any score taken individually. Using the already curated data of multiple DNA double-stranded break detection methods (such as CIRCLE-seq, GUIDE-seq, and others), we assessed five approaches for determining whether a guide has too high of an off-target risk: (1) Zhang score alone, (2) CFD score alone, (3) Zhang and CFD scores, (4) Zhang or CFD score, and (5) the average of the Zhang and CFD scores. 21
The AUROC for the Zhang and CFD scores were 0.86 and 0.89, respectively. When calculating the average of these two scores, the resulting measure outperformed both scores individually, with an AUROC of 0.92. We also assessed the performance of the machine learning algorithms, AdaBoost and Random Forest, when the Zhang and CFD scores were provided as model features. The machine learning models did not outperform the simple approach of calculating the average of the two scores. See Supplementary Figure S2 for the Receiver Operating Characteristic (ROC) curve, including Area Under the Curve (AUC) measures. As the average is faster to calculate and easier to interpret, this is the method implemented as the default option in Crackling. The four other approaches are also available as options in the configuration file.
Off-target scoring using ISSL
ISSL can be used to rapidly perform approximate nearest neighborhood searches in collections of locality-sensitive signatures. 18 By using fixed-length signatures as search keys, items in a neighborhood can be found in constant time. This approach was initially proposed as a high-performance method for searching web-scale collections of data. Given that genomic data are at a comparable scale, it provided a feasible method for evaluating CRISPR guide specificity.
The approach utilizes a bit-encoded index to reduce memory and storage requirements and to use CPU instructions most effectively. The four letters of the genomic alphabet (A, T, C, G) are 2-bit encoded; hence, a 20-bp genomic sequence requires 40-bits but is stored in a 64-bit word. In the context of Crackling, this 20-bp sequence represents an off-target site. For each bit-encoded signature, the critical 40-bit segment is portioned into
Separately, when considering guide specificity, the bit-encoding process is repeated for each candidate guide. Given that n mismatches are allowed and there exist
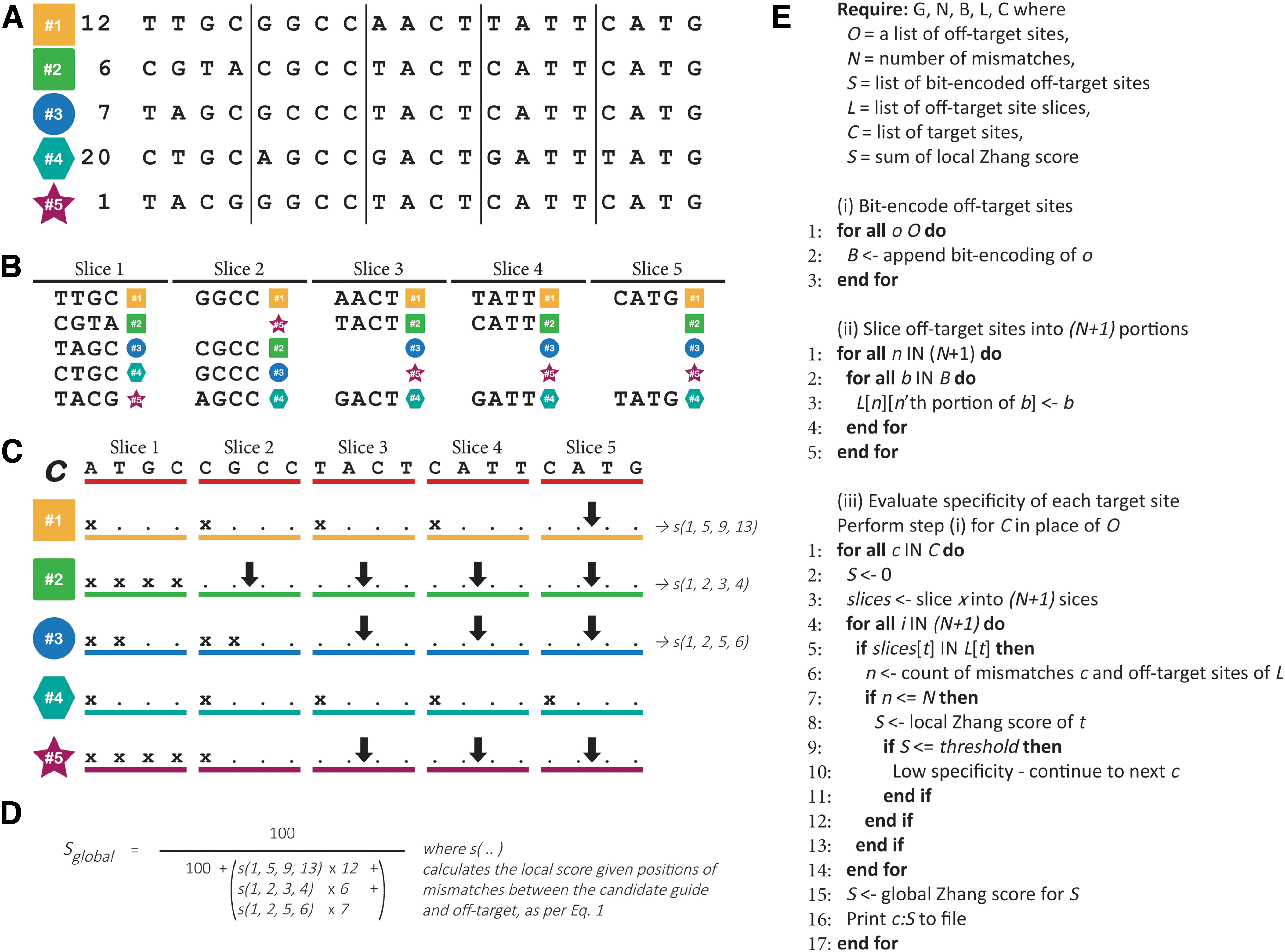
Off-target scoring performance
We timed each of the off-target tools on genomes of increasing size, which were obtained from the National Center for Biotechnology Information (NCBI) (Table 1). For each genome, candidate guides were extracted from the forward and reverse strands using a regular expression: [ACG][ATCG] 20 GG (and the complement for the reverse strand). Two editions of these target sites were created: one including the PAM sequence and one without, as this was required by the tools. This process was repeated for off-target sites, however, using the following expression: [ACG][ATCG] 20 [AG]G (again, with a complementing expression for the reverse strand).
The genomes used in this study
Asterisk indicates that these genomes were only used on the high-performance machine due to memory limitations.
Mb, megabases.
Using the candidate guides extracted for each genome, we further extracted 5 distinct sets of 10,000 candidate guides. For O. sativa, we repeated this but for sets of size 5000 to 25,000, incrementing by 5000. For each of these, we tailored copies for Crisflash and Cas-OFFinder, which required custom formats. ISSL required an index of the off-target sites. Crisflash required a single genome file, as opposed to a file for each chromosome. For this, we separated each chromosome with sufficient Ns to prevent false sites at the junction between chromosomes. FlashFry required a custom index for each genome but could not produce one for any genome larger than that of O. sativa.
For findMismatches, findMismatchesBit, and Crisflash, we modified the source code to time the guide specificity evaluation method in each tool. Additionally, for Crisflash, we made modifications to time the construction of its tree data structure. For Cas-OFFinder, we timed it with the time application with microsecond precision. For ISSL, we measured using the time library within Python. All preliminary tests were performed on a high-performance Linux workstation, with two 18-core Intel Xeon E5-2699 v3 (2.3 GHz) CPUs, 512 GB RAM, 4.2 GB allocated swap space, and Hewlett-Packard Enterprise MB4000GEFNA 4TB HDD. Further testing was performed on a different Linux workstation with one 8-core Intel Core i7-5960X (3.0 GHz) CPU, 32 GB RAM, 32 GB allocated swap space, and Samsung PM87 SSD.
For all tests, we used a strict 30-h wall time, as tools slower than this would not scale to the analysis of entire genomes.
Benchmarking guide design pipelines
We previously benchmarked leading CRISPR guide design methods using custom data sets derived from the mouse genome. 1 These data sets were of size 500,000 bp (500k), 1,000,000 bp (1m), 5,000,000 bp (5m), and the full 61m bp chromosome 19 (full). Some tools require an annotation file, and only consider guides in coding regions. To account for this, we calculated the effective base pairs per second (EFPS), which uses the length of the regions used for candidate extraction. Crackling considers the entire input genome; thus, the genome length was used when calculating the EFPS. We constructed the ISSL index for these data sets and ran identical experiments on Crackling from the original article.
Results
All preliminary tests were executed on a high-performance machine and followed up on a machine where the available memory was more limited. Cas-OFFinder could not be tested on the high-performance machine due to compatibility issues. For FlashFry, it was not possible to generate the required indexes for genomes larger than Oncorhynchus mykiss, and the tool was therefore not tested past this point. Generating the index for the larger genomes was attempted on both machines, with no success.
The ISSL approach powering Crackling is the fastest off-target scoring method
Three tools (findMismatches, findMismatchesBit, and ISSL) were capable of completing tests on all 12 genomes on the high-performance machine. They also completed tests on the 10 genomes considered on the workstation (with the 2 largest being excluded due to the memory limitation of this machine). On the high-performance machine, Crisflash failed on genomes larger than O. mykiss due to segmentation faults. On the workstation, it saturated physical memory, causing swapping to occur.
For both machines, the performance for findMismatchesBit was greater than findMismatches by a factor of up to 6 for all tests, confirming that bit-encoding alone provides a performance benefit. The results for the Triticum urartu data set on the workstation are an outlier due to memory being swapped to disk.
ISSL was able to evaluate 10,000 candidate guides for O. sativa in 12 s on average; whereas findMismatches completed analysis of the same data sets in an average of ∼7.5 min. This is a 38 × speedup. ISSL also significantly outperforms Cas-OFFinder. For instance, it completed the T. urartu test in ∼4 min on the workstation compared with 10 h for Cas-OFFinder. This is a 161 × speedup.
The mean run-times for each test are given in Table 2 and visualized in Figure 3A and B. ISSL is the fastest method for off-target scoring. For large genomes on low-memory machines, findMismatchesBit can also be a useful alternative, due to its lower memory footprint.

Run-time for an increase in genome size when using
Mean run-time, of five tests, when genome size is increased (min:s)
— Indicates the test failed due to memory limitations.
fM, findMismatches; fMb, findMismatchesBit; ISSL, Inverted Signature Slice Lists.
For ISSL and FlashFry, the supporting indexes containing off-target sites are generated in a preprocessing step that is required only once. For Crisflash, the tree data structure is constructed at run-time so we modified the source code to include a timer for this. We timed the construction of the indexes for each of these tools and report the results in Table 3 and Figure 3C and D.
Mean run-time for generating the off-targets index, of five tests, when genome size is increased (min:s)
— Indicates the test failed due to memory limitations.
Impact of the number of targets
We ran each tool on the O. sativa genome and varied the number of candidate guides to be evaluated. The results are shown in Table 4 for mean run-times over five tests.
Run-time when increasing the number of candidate guides to evaluate (min:s)
Tested on Oryza sativa.
ISSL completed all tests in less than 35 s, whereas Cas-OFFinder completed the test on the smallest sized data set in 23 min and largest sized data set in 2 h. Here, ISSL performed up to 300 × faster than Cas-OFFinder and approximately an order of magnitude faster than the next tool.
Crackling is a fast method to produce efficient guides
We constructed the ISSL index for the data sets generated in a previous article where we benchmarked CRISPR guide design pipelines. We repeated these experiments using Crackling, using its default settings. The consensus approach, by default, considers a guide as efficient when at least two of the three approaches agree. The optimization level, by default, is set to high, which, for each guide, progressively excludes tests to reduce run-time. The average EFPS over five tests for the data sets were:
500k: 11,012 EFPS
1m: 12,406 EFPS
5m: 22,487 EFPS
full: 34,440 EFPS
We also explored the effect on run-time when changing the consensus approach or optimization level, from their respective defaults to other options. Without changing the optimization level but adjusting the consensus approach, run-time can be reduced by making decisions early. For example, when accepting a guide when at least one of the three tools agrees, run-time is reduced, on average, by 16%. The strictest consensus approach, where all methods need to agree, meaning guides are rejected early, decreases run-time, on average, by 57%. Without changing the consensus approach, but adjusting the optimization level from high to low, run-time is increased, on average, by 84%. The low optimization level evaluates all tests for those guides that are seen only once during the initial discovery step. We recommend carefully considering the effects of changing the consensus approach as the precision of Crackling may change. If the user wishes to improve run-time, or to retrieve more detailed results, then modifying the optimization level is how this should be achieved.
The EFPS for Crackling, using the default configuration, is visualized in Figure 4. In the benchmark, we used this figure to define low-, medium-, and high-performance groups. Crackling enters the high-performance group and is the only method in that group to correctly filter guides. Two other tools in this group, CasFinder and CRISPR-ERA, reported scores. However, for both tools, the score was identical across all guides, and as such, it did not carry useful information. Compared with other tools that filter guides, Crackling scales to larger sized data sets the best.
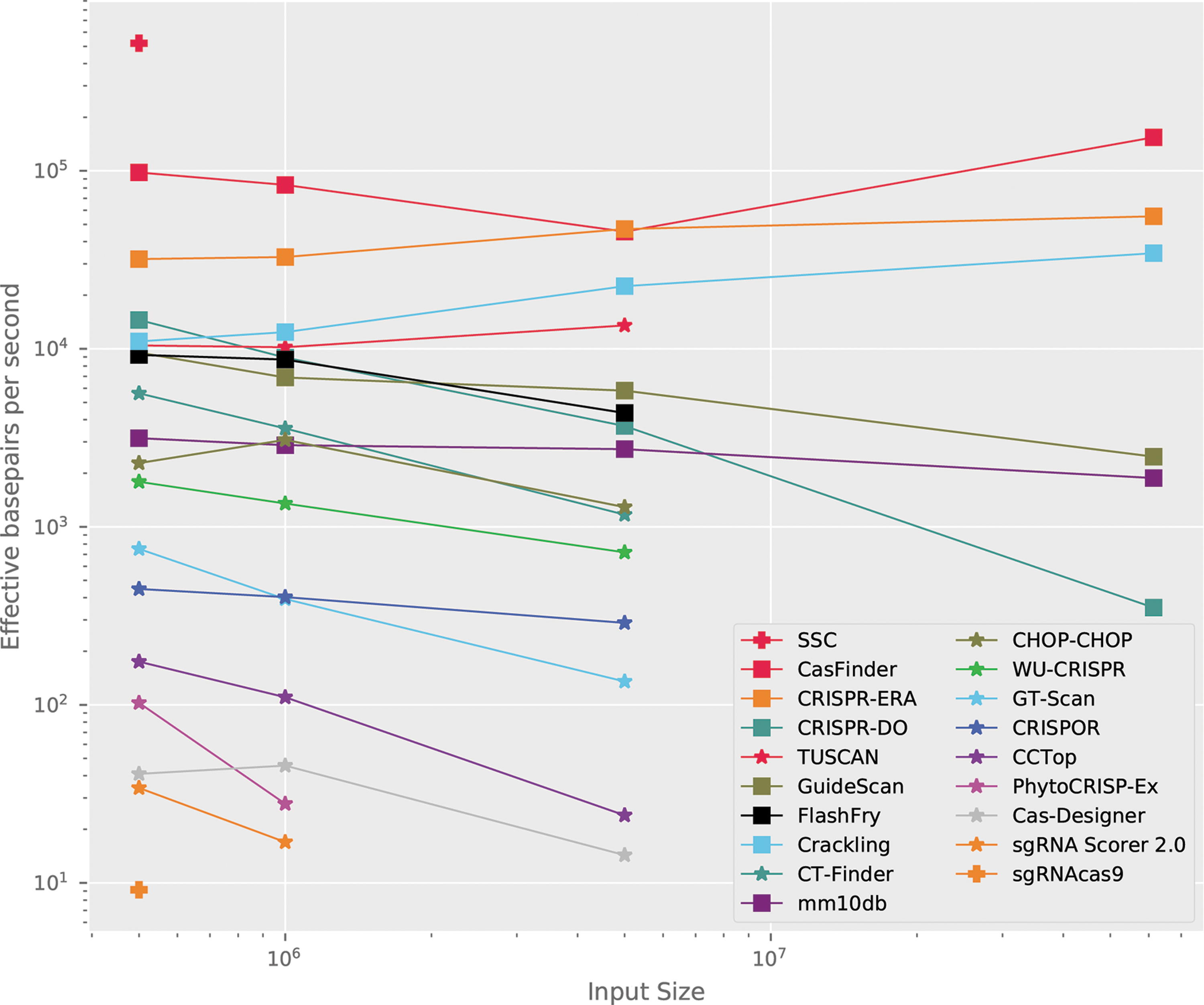
Run-time on mouse chromosome 19 data sets. This plot is extended from Bradford and Perrin 1 to include Crackling when ran with the default settings.
On the desktop workstation, we measured how fast Crackling was able to process guides for all genes in the human genome (for both on-target and off-target evaluation). It completed in 4 h and 36 min, without needing swap space for additional memory. This demonstrates the ability for Crackling to run on a standard workstation that does not have an unusually large volume of memory. In contrast, Crisflash is reported to need 90 GB of memory to process the human genome for off-target evaluation alone. 12 This volume of memory is usually available only in specialist computers. Even when we ran Crisflash on a machine with ample memory, it could not complete any of the experiments where the size of the input genome was close to that of the human (Table 2).
The quality of the guides produced by Crackling is also the highest among the tools reviewed. Table 5 describes the precision of each tool on two experimentally validated data sets, Wang and Doench.22,23 As shown in Figure 5A for the Wang data set, Crackling correctly selected many guides that are efficient and very few that are inefficient. The same result can be observed for the Doench data set in Figure 5B. Crackling, by default, requires a candidate guide to be accepted by two of the three scoring approaches to be selected. When reconfigured this to require all three approaches to agree, the precision of Crackling increases (to 90% and 48.5% on the Wang and Doench data sets, respectively), but the recall drops to values that are only acceptable when a low number of candidates is sufficient (7.4% and 8.9%, respectively).
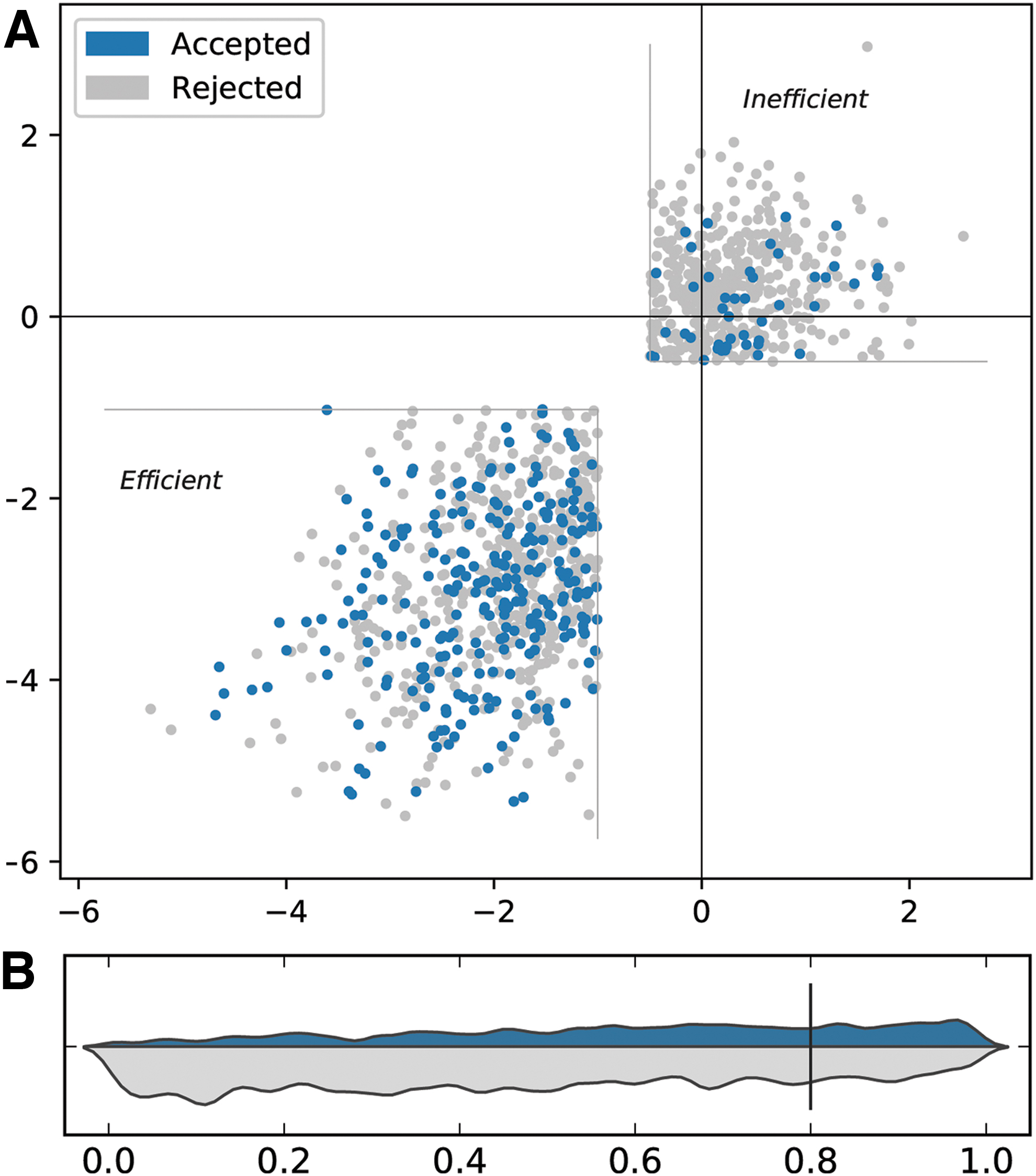
Results on the experimental data sets.
Precision of tools on experimentally validated data sets
The bolded configuration for Crackling is its default. Table was adapted from Bradford and Perrin. 1
— Indicates the tool was not tested on this data set as it was used for training.
Indicates the tool was not able to complete all benchmarking tests from Bradford and Perrin. 1
These data sets contained activity measures for guides that were expressed using U6 promoters. To explore the adaptability of Crackling to experimental conditions where T7 promoters are used, we used a data set, as prepared by the authors of the CRISPOR pipeline, that contains mutation rates for 107 guides expressed using T7 promoters. 7 The distribution of mutation rates suggested that a threshold of 0.2 is suitable to classify guides as being efficient or not. Using the default configuration of Crackling, a similar F1 score was obtained (57% compared with 51.4% for the Wang data set). However, this is with a lower precision (65%) and higher recall (52%). These results are preliminary, as publicly available data using T7 promoters appear to be sparse, but they suggest a possible impact. We will continue to investigate this as more data become available. The modularity and consensus-based approach that characterizes Crackling allows the future inclusion of additional scoring methods to address this if needed.
Discussion
One of the main challenges of experiments using CRISPR-Cas9 systems is the design of suitable guide RNAs. It is crucial, but not trivial, to ensure that these guides will be efficient but also specific enough not to lead to off-target modifications. Currently, there are no tools that meet both objectives while remaining practical to use on large genomes. In this article, we presented Crackling, a new tool for whole-genome identification of suitable CRISPR targets. Crackling is powered by a new high-performance off-target scoring approach based on ISSL and utilizes our previously published consensus approach for improving the precision when predicting guide efficiency.
When executing ISSL on a set of genomes using a high-performance machine, ISSL performs approximately an order of magnitude faster than the next best performing tool (findMismatchesBit), and up to two orders of magnitude faster than the worst performing tool (findMismatches).
We ran a number of off-target scoring tools on the O. sativa genome and varied the number of candidate guides to be evaluated. It would be expected that run-time is a linear function of input size, seeing that each candidate guide is evaluated individually. For all tools, we find that this is true.
Overall, our findings are that Cas-OFFinder is the poorest performing tool; findMismatches and findMismatchesBit are proportional; ISSL is the best performing, followed by Crisflash and finally FlashFry.
The consensus approach, which by default only accepts guides that have been predicted as efficient by at least two of the three scoring methods, provides gains in precision while preserving a reasonable recall. Changing the configuration of the consensus approach can further improve the precision through sacrificing recall. The precision, recall, and F1 score for all configurations are shown in Table 5.
Taken together, all these results show that Crackling can produce high-quality results in a relatively short period. It is among the best performing tools with respect to both critical properties (specificity and efficiency) that are considered when evaluating CRISPR guides.
We show that it provides the fastest way to calculate off-target risk, by using binary encoding and the ISSL method to identify closely related sites throughout the genome. This is confirmed on 12 genomes of increasing length. We also show that by combining the results of multiple scoring approaches, we can maximize the efficiency of the guides being designed. On experimental data, the set of guides it selects is better than those produced by existing tools.
Overall, this makes Crackling a faster and better method to design guide RNAs at scale.
Crackling is available as a standalone pipeline via GitHub (https://github.com/bmds-lab/Crackling) and will run on any modern workstation. For unusually large genomes, a machine with extra memory may be required. Installation and usage instructions are provided in the repository. Support is available by submitting a report using GitHub Issues.
Footnotes
Acknowledgments
We would like to thank the Big Data laboratory at the Queensland University of Technology for providing access to their high-performance machine, and the authors of the other tools for making their source code available.
Authors’ Contributions
J.B. and D.P. designed the study, analysed the results and wrote the manuscript. J.B. and D.P. implemented the on-target evaluation. J.B. and T.C. implemented the off-target scoring. All authors have reviewed and approved the article before submission. This article has been submitted solely to The CRISPR Journal, is not published, nor in press.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
J.B. was supported by the Australian Government Research Training Program Scholarship. D.P. is supported by the Australian Research Council (ARC Discovery Project DP210103401).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.