
Abstract
Ribonucleoproteins (RNPs) are frequently applied for therapeutic gene editing as well as fundamental research because the method is fast, viral free, and shows fewest off target effects. We evaluated various parameters to genetically engineer human hematopoietic stem and progenitor cells (HSPCs) using Streptococcus pyogenes Cas9 (spCas9) RNPs, and achieve gene editing efficiencies up to 80%. We find that guide RNA (gRNA) design is critical to achieve high gene editing efficiencies. However, finding effective gRNAs for HSPCs can be challenging, while the contribution of numerous in silico models is unclear. By screening more than 120 gRNAs, our data demonstrate that in silico gRNA prediction models are ineffective. In this study, we established a time- and cost-efficient in vitro transcribed gRNA screening model in K562 cells that predicts effective gRNAs for HSPCs. RNP based screening thus outperforms in silico modeling and we report that gene editing is equally efficient in distinct CD34+ HSPC subpopulations. Furthermore, no effects on cell proliferation, differentiation, or in vitro hematopoietic lineage commitment were observed. Finally, no upregulation of p21 expression was found, suggesting unperturbed HSPC homeostasis.
Introduction
Genome editing is a powerful tool to study gene function and correlation, and the fast turnover time of generating knockout cells makes this technique already indispensable for most research fields. Various genome editing technologies have been developed in the past decade, including zinc-finger nucleases,1,2 transcription activator-like effector nucleases,3,4 and the RNA-guided Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated (Cas) systems.5,6 A variety of Cas proteins from multiple species have been identified, 7 with Streptococcus pyogenes Cas9 (spCas9) and modifications thereof being the most developed gene editor to date with several updates that provide distinct features.8–12
In mammalian cells, spCas9 and single guide RNAs (sgRNAs) were initially co-expressed using transfected plasmid DNA in immortalized cells followed by clonal selection. 13 Despite its success in immortalized cells, clonal selection hampers the applicability to use gene editing in primary human hematopoietic stem and progenitor cells (HSPCs) as the expansion potential on single clones is limited both in vitro and in vivo. Highly efficient gene editing on bulk HSPCs can potentially overcome these limitations and can make gene editing an indispensable tool for fundamental and translational research, as well as therapeutic purposes. Nonetheless, genome editing in primary hematopoietic cells requires experimental optimization as efficiencies are target specific. 14
Previously, several optimization steps were made, for example, direct spCas9 protein delivery, which outperforms spCas9 delivered as mRNA or expressed through plasmid DNA. 15 In addition, various spCas9 modifications revolve around optimizing nuclear localization signals (NLS), which can improve the delivery of RNPs to the nucleus.16,17 RNP delivery can be performed using iTOP 18 or more commonly reported by nucleofection.14–19 For nucleofection, various buffers have been reported that potentially could improve efficacy or reduce the costs for RNP-based delivery on a large scale.14,20
For RNP-based gene editing, gRNAs were initially in vitro transcribed (IVT). However, chemically synthesized gRNAs, lacking the 5′triphosphate cap, 15 outperform in vitro transcribed single gRNAs (IVTsgRNAs) by preventing RIG-1-mediated intracellular immune responses in primary cells, thereby leading to fewer cellular off-target effects.21,22 Moreover, chemically synthesized RNAs contain modifications that improve RNA stability and have been shown to be more effective compared with IVTsgRNAs. 15
Despite these improvements, gRNA efficacy must be determined empirically in HSPCs, as it is recommended to test five to eight gRNAs for each target, 14 suggesting that most gRNAs are ineffective. Numerous in silico-based algorithms were developed to predict on-target gRNA efficacy23,24 and can be used to aid in gRNA selection to genetically modify hematopoietic cells. However, it is unclear how these algorithms translate to RNP-based delivery and which algorithm is most accurate to predict activity in HSPCs.
CRISPR-spCas9-induced double-strand breaks in HSPCs can potentially lead to impaired proliferation and differentiation as previously shown for retinal epithelial cells, human pluripotent stem cells, and HSPCs.25–27 Importantly, DNA damage in hematopoietic stem cells, coupled to failure or faulty of repair, can lead to pathology, premature senescence, bone marrow failure, and myeloproliferative neoplasms.28,29
In this study, we evaluated various parameters that need to be considered for genomic editing in hematopoietic cell lines and human HSPCs including spCas9 source, nucleofection conditions, and gRNA design. To determine the most optimal gRNA criteria to genetically modify HSPCs, we have to setup a fast IVTsgRNA screening protocol in the K562 cell line that more closely relates to the hematopoietic system compared with previously reported algorithms.23,30–37 By testing >120 different gRNAs using RNP-based delivery, we demonstrate that IVTsgRNA screening in K562 is effective to predict RNP-based genome editing in human HSPCs and outperforms in silico gRNA selection tools that are currently available. From the 10 in silico algorithms that are available, we find that the Doench 30 and Wang 37 scores are most accurate to predict effective gRNAs to genome edit HSPCs but inferior to the cellular screening method shown here. RNP-induced gene editing did not lead to p21 upregulation or HSPC proliferation or differentiation defects.
Materials and Methods
All reagents and resources are described in Supplementary Table S1–S6 as well as in the Supplementary Materials and Methods.
spCas9 protein expression and purification
spCas9-3xNLS (plasmid no. 114365; Addgene) was expressed in Bl21 star cells (no. C601003; Thermo Fisher) and purified in a two-step purification method (HisTrap, followed by size exclusion). Protein concentration of spCas9 concentrations was determined in bicinchoninic acid assay (BCA) proteins assay and snap frozen at −80°C (see the Supplementary Data for more detailed protocols and resources).
IVTsgRNA synthesis
gRNAs were designed using CRISPOR (Version 4.98) by selecting gRNA sequences with the highest specificity score (unless stated otherwise). crRNA sequences were developed as ssDNA oligo's including a T7 promoter and an overhang sequence resulting in a 56–58 nucleotide oligo (see Supplementary Table S4, e.g., note that 1 or 2 guanines could be added in some cases to boost transcription but might also change the outcome of efficacy). This oligo was annealed and amplified to a fixed 91nt oligo containing the scaffold domain using Taq polymerase (cat. no. 10342046; Invitrogen). Transcription was performed using HiScribe T7 (E2040; NEB) and purification with monarch columns (T2040; NEB). Concentration was measured using NanoDrop (see the Supplementary Data for more detailed protocols and resources).
High-throughput IVTsgRNA screening
Three micrograms of spCas9-3xNLS +1 μg IVTsgRNA was resuspended in 20 μL homemade nucleofection buffer 14 using 0.2 × 106 K562 cells. Twenty microliters of the RNP-K562 cell mixture was nucleofected using FF-120 (Amaxa 4D), and 80 μL of culture media was added directly after transfection. Without washing, the complete 100 μL nucleofection mix was transferred and cultured in 24-well plates and cultured in 500 culture media. The complete process of RNP formation, transfection, and washing of the strips was performed using a multichannel and takes ∼2–3 h to transfect 48 gRNAs.
Note that nucleofection strips and cuvettes can be reused as described previously. 14 At least 30,000 cells were used for DNA isolation in high throughput (HT)-vacuum manifold according to the manufacturer's conditions (MN 740455.4) that can process up to 96 samples in 1 h. Two microliters of DNA isolate was used for polymerase chain reaction (PCR) (primers are shown in Supplementary Table S4), and PCR cleanup was performed using Exo-SAP (M0293, M0371; NEB). Sanger sequencing reactions were prepared using (no. 4337455; Thermo Fisher). Tracking of Indels by DEcomposition (TIDE) analysis 38 was used to determine the indel frequency.
CD34 isolation and nucleofection
Human material was obtained after informed consent, mobilized peripheral blood (MPB) was obtained from leukapheresis material, and cord blood was collected according to the guidelines of NetCord FACT (Sanquin Cord Blood Bank, The Netherlands). Cryopreserved CD34+ cells were thawed and cultured in Cellquin media, 39 supplemented with a cytokine maintenance cocktail containing stem cell factor (SCF) (100 ng/mL), Fms Related Receptor Tyrosine Kinase 3 ligand (FLT3LG) (100 ng/mL), and thrombopoietin (TPO) (10 ng/mL). All cells were precultured for 16–30 h before nucleofection. CD34 cells were collected and resuspended in nucleofection buffer from Amaxa or homemade M1 buffer 14 and nucleofected using program EO100 unless otherwise stated.
Directly after nucleofection, room temperature culture media were added and cells were spun to remove residual nucleofection buffer. Cells were cultured with Cellquin media including interleukin (IL) 3 (100 ng/mL), IL6 (100 ng/mL), TPO (10 ng/mL), and SCF (100 ng/mL) at 37°C, 5% CO2. Expansion and proliferation were performed as previously described. 39 Cell survival was determined using flow cytometry and quantified for viable cells relative to control cells using Live/Dead Near-IR Dead Cell Staining Kit (L34975; Invitrogen).
RNP formation for synthetic gRNAs used in HSPCs
Synthetic crRNAs (Alt-R; IDT) and synthetic scaffold RNA (both from IDT, DNA Technologies) were dissolved as 100 μM stocks using ultra-pure RNAse-free water. From this stock, 2.25 μL crRNA was incubated with 2.25 μL Tracer RNA and incubated at 95°C for 5 min. Next, the mix was incubated at room temperature (RT) for 20 min to allow gRNA formation. spCas9 was added and incubated with gRNA at RT for 10 min to form RNPs. For 1 RNP unit, 100 pmol Cas9 + 225 pmol gRNA, as previously reported, was used. This is similar to 15 μg Cas9 and 6 μg RNA. For 2 RNP units, 30 μg Cas9 + 450 pmol gRNA was used, and for 3 RNP units, 45 μg Cas9 + 675 pmol gRNA was used. 14 All gRNA sequences are reported in Supplementary Table S1–S4. gRNA sequences targeting CD33 and CD45 were from Ting et al 40 and CD44 from Liu et al. 41
In vitro cleavage assay
A PCR product of hCD45 was generated (primers in Supplementary Table S4) and purified using a PCR purification kit (GE28-9034-70; GE healthcare). One hundred nanograms of PCR template was incubated in NEB buffer 3.1 for 1 h at 37°C with indicated amounts of preformed RNPs (gRNA, see Supplementary Table S1). Next, PCR products were incubated with 2 μg/μL Prot K and hence analyzed using gel electrophoresis. PCR bands were quantified using the ImageJ software.
Cell line culture and nucleofection
K562, EOL-1, NB4, and HL60 were cultured in Roswell Park Memorial Institute (RPMI) medium (130-046-703) containing 10% heat-inactivated fetal bovine serum (FBS), whereas ML2 was cultured in RPMI medium containing 15% and 20% FBS. All cell lines were nucleofected with homemade nucleofection buffer. 14 NB4 and HL60 were nucleofected using program X-01, ML2, and EOL-1 at CA-137 and T-016 for K562. For HEK293, we used Amaxa SF cell line kit with Amaxa-recommended program CM-130.
GFP competition assays
Cell lines were transduced using RetroNectin (cat no. T202; Takara Biosciences) according to the manufacturer's conditions. The GFP reporter was a kind gift from Kosuke Yusa (plasmid no. 67980; Addgene), and BFP+ cells were enriched using fluorescence-activated single cell sorting (FACS), in case transduction efficiency was <90%. Transduced cells were mixed 1:1 with their wild-type counterparts before nucleofection, and the BFP ratio was determined on cells that were not transfected with 5–10 μg Cas9-3xNLS.
Western blot and flow cytometry
Lysates for western blot were made in a 1% Triton buffer including complete protease inhibitor cocktail (11697498001; Roche) and western blot and flow cytometric staining were performed as described previously.42,43 All antibodies for flow cytometry and western blot can be found in Supplementary Table S5. For the p21 staining, we used the exact protocol as described by Cell Signaling Technologies. Briefly, at least one million cells were stained with Near-IR 1:200 L/D staining and hence fixed in 4% paraformaldehyde for 15 min at RT. After washing with phosphate-buffered saline (PBS), cells were permeabilized with ice-cold 100% methanol for 30 min on ice. The cells are stored at −20°C or directly used. After washing with PBS, a staining with p21 antibody (Supplementary Table S5) (1:50) was performed for 1 h at RT, protected from light. After washing, cells were resuspended in PBS/0.5% bovine serum albumin and measured using flow cytometry.
Results
Efficient gene editing in K562 is dependent on spCas9 NLS variation and nucleofection buffer
To setup a model system for IVTsgRNA screening, we started by optimizing gene editing in the hematopoietic cell line K562. spCas9 purification was performed (Supplementary Fig. S1A–F), and the commercially available cell line kit, suited for K562 cells, was compared with the previously reported homemade K562 cell line solution. 14 K562 cells were nucleofected with an ATTO-labeled ribonucleoprotein (RNP) complex targeting CD33. Nucleofection using this homemade solution resulted in significant higher transfection of RNPs (Fig. 1A) and increased knockout efficiencies with no significant differences in cell survival (Fig. 1B), compared to the commonly used Amaxa kit. Overall, this leads to significant different nucleofection scores that take survival and knockout into account (Fig. 1B, right panel). Importantly, the homemade buffer is low cost and thereby ideal for high-throughput applications or bulk experiments and can be stored for at least 12 weeks without loss of activity (Supplementary Fig. S1G).
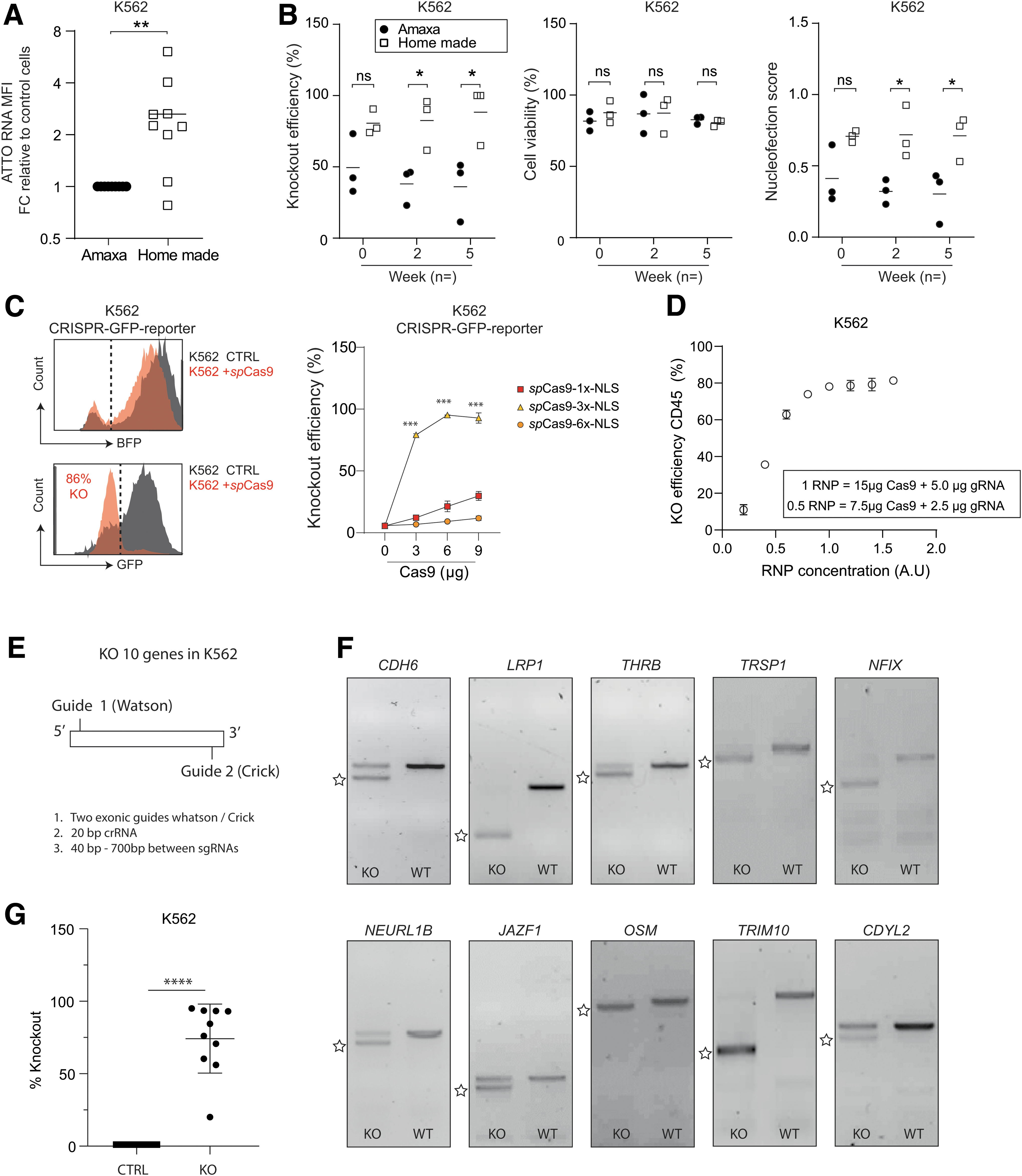
Efficient gene editing in K562 is dependent on spCas9 NLS variation and nucleofection buffer.
Next, we evaluated recombinant spCas9 proteins with different NLS sites. NLS number and sequence can substantially influence genome editing efficiencies.16,17 Therefore, K562 cells were lentivirally transduced to stable co-express a bicistronic BFP-GFP reporter and gRNA against GFP (Fig. 1C). All transfected spCas9 proteins generated knockouts in a dose-dependent manner. However, 3xNLS-spCas9 17 is significantly more potent compared with 1xNLS 44 or 6xNLS, 16 while cleavage activity and transfection efficiency were not different (Supplementary Fig. S1H, I). This indicates that fusion of specific nuclear localization motifs can significantly enhance genome editing efficiencies. Of note, knock out (KO) efficiencies for 3xNLS-spCas9 reached a plateau in K562 between 0.6 and 1.0 RNP units (Fig. 1D).
We next determined if this in-house generated low-cost RNP nucleofection protocol is suitable for high-throughput target validation. For 10 genes, 2 gRNAs on opposite strands were designed (Fig. 1E and Supplementary Table S1), which has previously been shown to enhance genome editing efficiency. 45 Strikingly, we find that 9/10 conditions show significant gene alterations, with efficiencies ranging between 50% and 100% (Fig. 1F, G), providing evidence that this RNP delivery system is highly suitable for fast and efficient target validation of multiple genes.
Highly efficient genome editing in human HSPCs is mainly dependent on gRNAs design
Encouraged by the results in K562, we subsequently aimed to evaluate the translation for genome editing in human HSPCs and therefore assessed various nucleofection conditions. Several programs are currently reported to transfer RNPs into HSPCs;14,17,40 however, it is unclear which program works best. Here, we confirmed that nucleofection program EO100 works significantly better compared with other previously reported programs, indicated by the nucleofection score that takes both survival as well as transfection efficiency into account (Fig. 2A). Next, it was determined which nucleofection buffer is superior to transfer RNPs into HSPCs, using a previously validated gRNA targeting CD4540 in combination with spCas9-3xNLS.
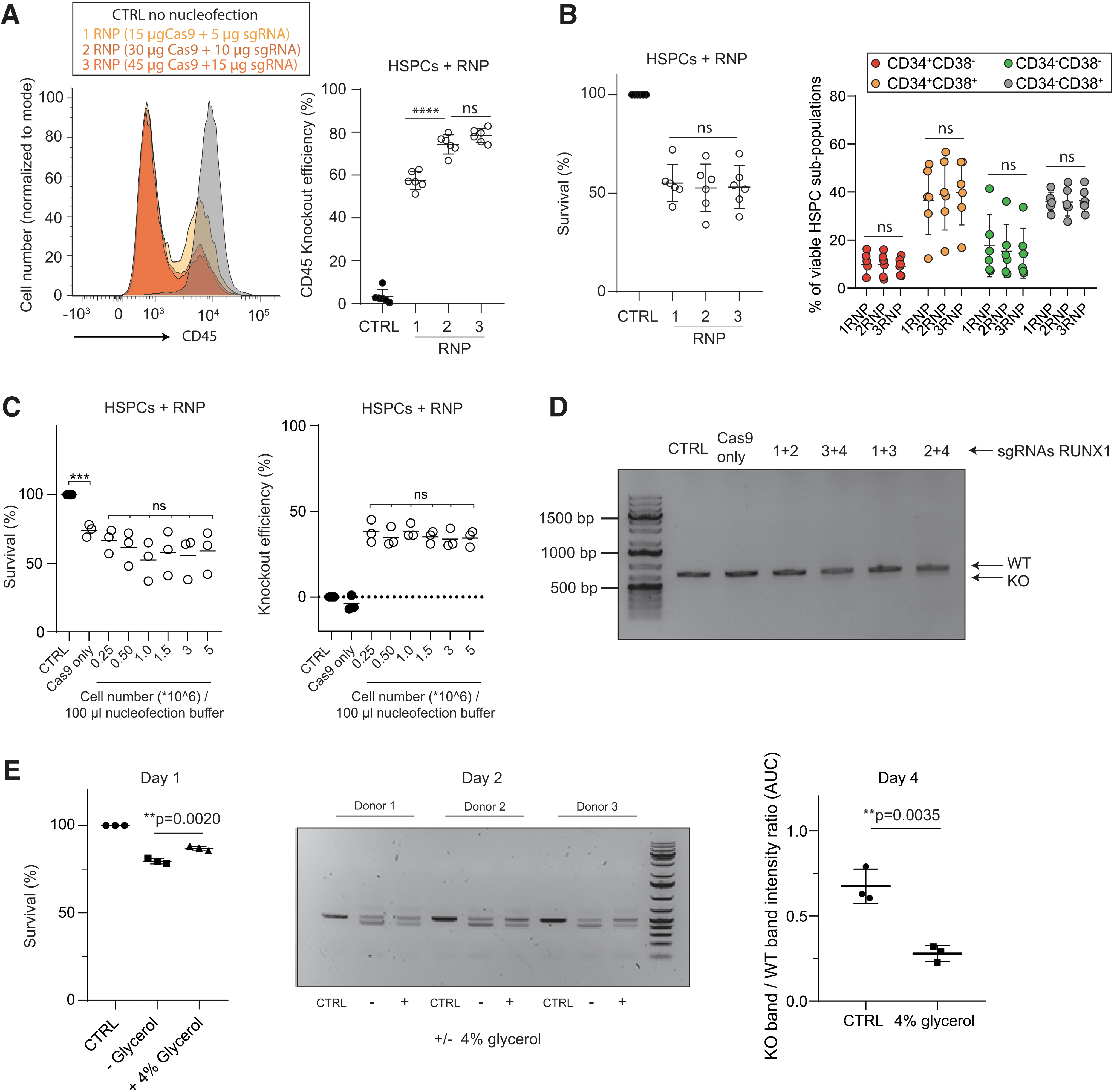
Highly efficient genome editing in human HSPCs is mainly dependent on gRNAs design.
Of note, we validated that spCas9-3xNLS outperforms spCas9-1xNLS in HSPCs (data not shown). This revealed that Amaxa P3 provides significant higher gene editing efficiencies compared with previously reported buffers14,20 (Supplementary Fig. S2B); however, differences were generally small, and homemade nucleofection buffers might alternatively offer an advantage to maintain cost-efficiency. Then, it was evaluated which RNP concentration is most optimal to gene edit human HSPCs using CD45 targeting RNPs on distinct HSPC donors. This experiment revealed that two to three RNP units is most efficient to achieve high gene editing activities in HSPCs (Fig. 2A), while cell viability and immunophenotypes of different HSPC subsets remained similar (Fig. 2B).
Surprisingly, no effect on cell survival or gene editing activity was observed when different cell numbers were targeted with a fixed amount of RNP molecules (Fig. 2C). Together, these data suggested that high gene editing efficiencies can be achieved in HSPCs. Then, it was tested if similar gene editing activities could be reached using other gRNAs. Here, we selected four gRNAs to target RUNX1 and tested these gRNAs in pairs on HSPCs (Fig. 2D). We observed that gene editing efficiencies were generally low and agree with previous findings that most gRNAs are ineffective in HSPCs using RNP delivery. 14 Adding glycerol to the RNPs, increases survival of HSPCs as previously suggested, 17 however, also a dramatic decrease in gene editing efficiency was observed (Fig. 2E).
IVTsgRNA K562 screening system accurately predicts gRNA activity for HSPCs
Because most gRNAs are ineffective in HSPCs using RNPs, effective gRNAs need to be identified by either screening, or in silico modeling. Screening with synthetic gRNAs is expensive and affects the stocks of precious CD34+ HSPC resources. While the contribution of numerous in silico models is unclear, as none of these algorithms are validated for RNP-based gene editing in HSPCs.
To overcome this problem, we developed a cost-efficient and rapid IVTsgRNA screening assay in hematopoietic K562 cells. We hypothesized that this model is more relevant to identify effective gRNA than in silico modeling, as in silico models are mainly based on lentivirally expressing gRNAs in epithelial cells (summarized in Supplementary Table S1). IVTsgRNAs can be screened in bulk as they are low in costs and are a potential match with K562 cells that previously showed no activation of the RIG-1 pathway. 21
For 6 genes, 5–10 gRNAs were designed (Supplementary Table S2), and PCR templates including a T7 promoter were generated and used to express IVTsgRNAs (Supplementary Fig. S3A–C). Screening with this panel of IVTsgRNAs in K562 revealed for most genes at least one candidate that potentially predicts effectivity in HSPCs (Fig. 3A). To test our hypothesis, the efficacy of the top performing gRNA candidates was then monitored in primary HSPCs using hybridized synthetic gRNAs (Fig. 3B). This revealed an average indel score of 37%, and we concluded that this IVT screening system in K562 can be used to identify effective gRNAs to gene edit HSPCs. Note that synthetic gRNAs were used for HSPCs as IVTsgRNAs upregulated the RIG-1 pathway and induce differentiation and or apoptosis in primary hematopoietic cells (data not shown).
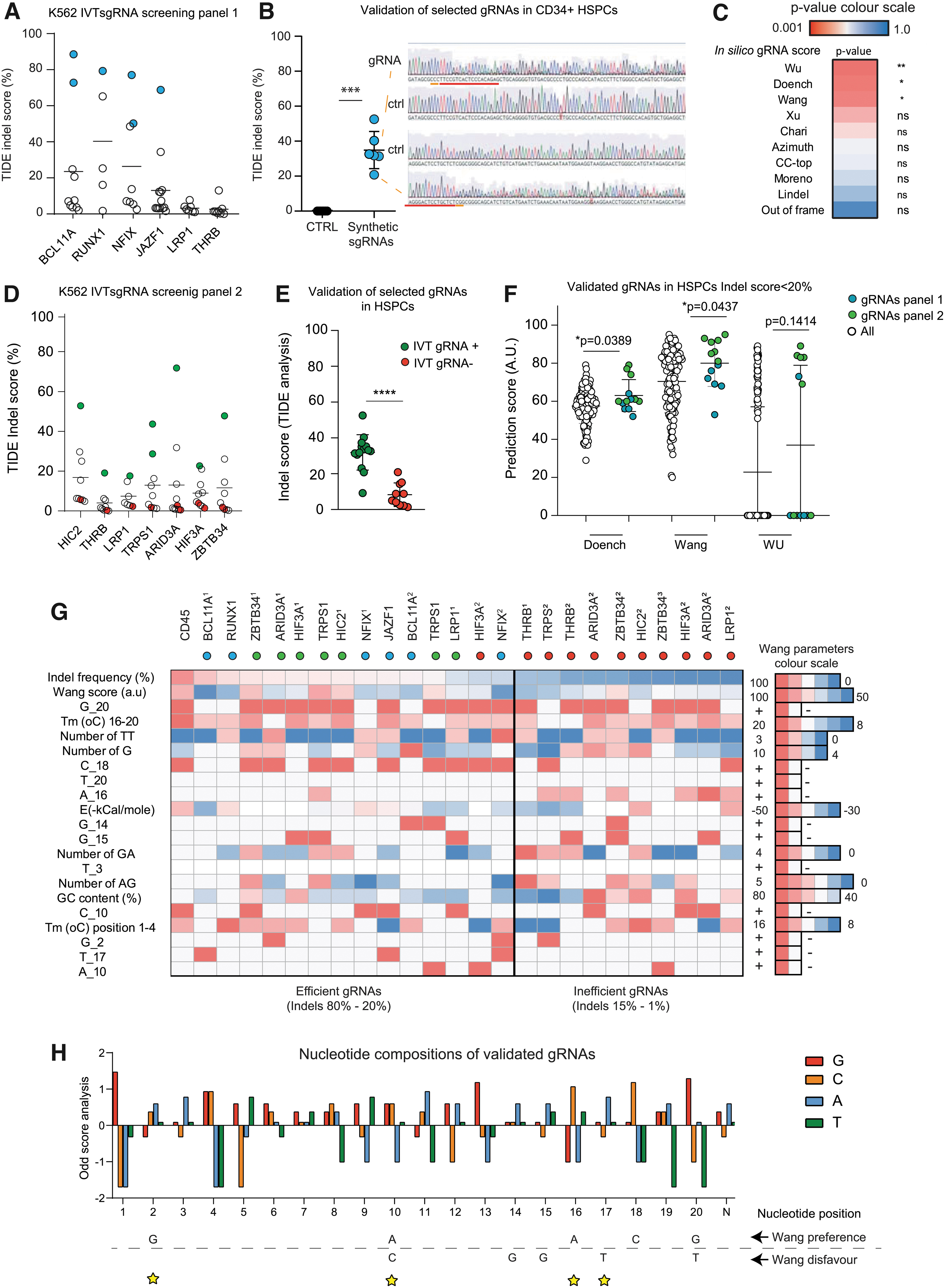
IVTsgRNA K562 screening system accurately predicts gRNA activity for HSPCs.
With this panel of validated gRNAs, we addressed the question which in silico algorithm would retrospectively have predicted most accurately these validated gRNAs (Supplementary Fig. S3D). Here, we identified Wang, Wu, and Doench’16 score as most predictive to enrich for gRNAs that are effective in HSPCS (Fig. 3C).
Based on these observations, a second panel of IVTsgRNAs was designed; this time IVTsgRNAs were selected with high Doench–Wang and Wu scores (Supplementary Table S3). Screening in K562 revealed several potential effective gRNAs (Fig. 3D). Given that most of the gRNAs are ineffective, we hypothesized that either in silico prediction scores are ineffective or that the IVTsgRNA screening model results in false negatives. For this reason, top candidates (green) were validated in HSPCs (Fig. 3E); however, this time also gRNAs were tested in HSPCs with a similar Doench, Wang, and Wu score that were found ineffective in the K562 IVTsgRNA screening model (red). The gRNAs identified in the K562 screening showed a significant higher indel score in HSPCs compared with their gRNAs that were assigned as ineffective.
This proves that screening of IVTsgRNAs in K562 is a predictive model for using synthetic gRNAs in HSPCs. To find which algorithm would have been retrospectively most effective, all validated gRNAs with an indel score higher than 20% were plotted and compared with the complete data set (Fig. 3F). This analysis revealed that the Wang score is most predictive; however, given that effective gRNAs can be found within the complete range distribution of the Wang score, we conclude that cellular screening is still superior. No correlation was found between the Wang score and indel formation (Supplementary Fig. S3E). In search for an explanation why solely in silico modeling is not sufficient, the strongest parameters from the Wang score were analyzed for the validated gRNAs and compared with their counterparts that were ineffective (Fig. 3G).
Moreover, the exact sequences of the validated gRNAs were aligned (Supplementary Fig. S3F) and clustered in three groups with different Wang and indel scores. The specific bases at position 17, 18, and 20 are critical in the Wang score, 37 and a similar dependency is observed in all gRNAs from the cluster-I Wanghigh/IndelHigh (Supplementary Fig. S3F). Interestingly, most gRNAs that we identified with random screening (cluster-II, Wanglow/HighIndel) lack these preferences and thereby partly explain the ineffectivity of in silico modeling. To find potential other features that might explain differences in gRNA screening and modeling, we combined all the efficient gRNAs that were validated in HSPCs (cluster I and II) and calculated Log Odds scores (Fig. 3H). Multiple differences were observed on our validated set versus the Odds scores as calculated by the Wang algorithm. Based on this analysis, we failed to recognize a trend for some of these Wang superpositions that are included in the algorithm (Fig. 3H).
For instance, our data set shows the absence of an adenine on position 16 in the effective gRNAs (cluster I and II), while abundantly present in the ineffective gRNAs (Fig. 3H and Supplementary Fig. S3). Furthermore, we observed that a guanine on position 1 is more preferred than a guanine on position 2. To find if the efficacy of IVTsgRNA screening is cell type specific, gRNAs from panel II were transfected into HEK293 cells and analyzed for indels (Supplementary Fig. S3G). This revealed target-dependent significant differences. To explore if the addition of one or two guanines, which we used to boost gRNA expression, 46 had an effect on potential decreased IVTsgRNA efficacy, the validated gRNAs were for their match and mismatch frequency (Supplementary Fig. S3H). This revealed that most identified gRNAs have a mismatch on at least one position and suggest that the addition of G or GG has no or limited inhibitory effect on gRNA performance.
Moreover, we also compared the efficacy of IVTsgRNAs with mismatches on these positions in K562, and here, we observe in one of two cases that the gRNA efficacy is substantially inhibited by the addition of GG (Supplementary Fig. S3I). This might suggest that screening with boosted expression may result in finding less effective gRNAs. For this reason, the algorithms were reanalyzed; however, this time all the gRNAs where GG was added and showed indel scores lower than 10% in K562 were removed from the analysis. This reveals similar results that most in silico models are not predictive (Supplementary Table S6 and Supplementary Fig. S3J). In conclusion, we find that IVTsgRNA screening in K562 is more accurate to predict gRNAs efficacy in HSPCs compared with in silico modeling. For preselection of IVTsgRNA screening, we recommend to use the Wang and Doench algorithm and we speculate that more effective gRNAs might be found in case the guanines are not added upstream the first nucleotide from the gRNA.
Genome editing efficiencies are equally represented within distinct human HSPC subpopulations
HSPCs can be divided in different subpopulations with distinct self-renewal and/or differentiation properties. 47 Knockout efficiencies in CD34+CD38− immature hematopoietic stem cells, CD34+CD38+ HSPCs, and CD34−CD38+ myeloid lineage committed cells were similar (Fig. 4A, B). Further subdivision of the CD34+CD38− cells using CD45RA and CD90 into immature hematopoietic stem cells48,49 demonstrated that genome editing efficiency was similar to downstream progenitors, although that events assayed were rather low (between 150 and 4200; Supplementary Fig. S4). Overall, we show that genome editing of HSPCs is consistent between donors and that genome editing efficiencies are similar between CD34+ HSPC subpopulations.
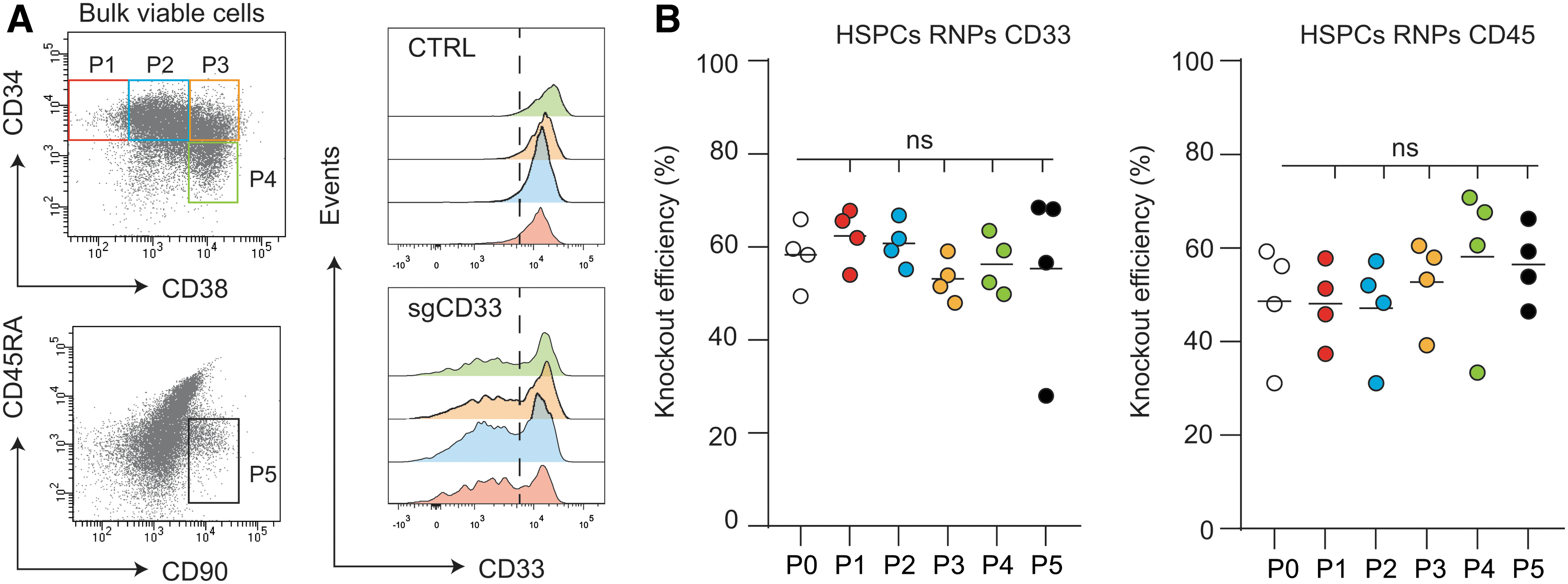
Genome editing efficiencies are equally represented within distinct human CD34+ HSPC subpopulations.
p21 is not induced after highly efficient genome editing in human HSPCs
CRISPR-spCas9 induces double-strand breaks, which has been reported to lead to p53 DNA damage response pathway activation and transcriptional activation of cyclin-dependent kinase inhibitor p21CIP1/Waf1 regulating G1 cell cycle progression. Genome editing may potentially lead to enrichment of cells that are, to a variable degree, defective in their p53 response.25–27
For this reason, we sought to determine if human HSPCs upregulated p21 when targeted with spCas9 RNPs. Incubation with Nutlin, an inhibitor of the interaction between MDM2 and p53, thus stabilizing p53, leads to a stark decrease in cell survival in wild-type p53 (p53wt) AML cell lines but not in p53 mutant cell lines (Fig. 5A). Nutlin induced p21 expression specifically in p53wt cells but not in p53 mutant cells indicated by western blot (Fig. 5B
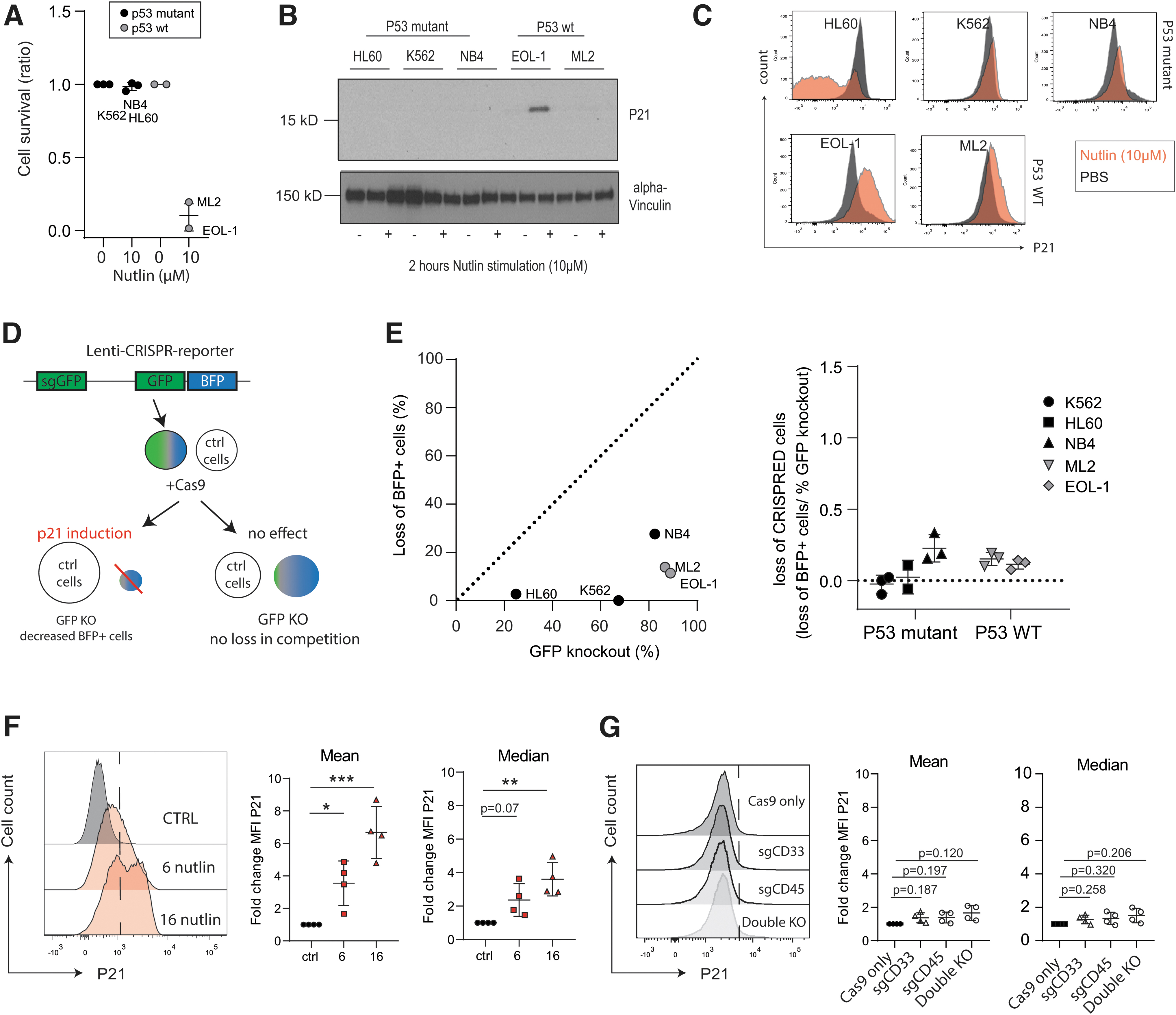
p21 is not induced after highly efficient genome editing in human CD34+ HSPCs.
Note that ML2 is weakly positive, indicated by an increased intensity image (Supplementary Fig. S5A). Induction of p21 was confirmed by flow cytometry and expression levels correlated with protein levels (Fig. 5C). Next, we assayed if CRISPR-induced Double Strand Breaks (DSBs) in p53wt cells lead to reduced cell survival as observed upon stabilizing p53 by Nutlin. The bicistronic BFP-GFP-gRNA CRISPR reporter gene was stably expressed in the five different cell lines (three p53wt and two p53 mutant lines; Supplementary Fig. S5B). Of note, nucleofection programs were optimized for all indicated cells (Supplementary Fig. S5C–E). The BFP-GFP-CRISPR reporter lines were co-cultured with their wild-type counterparts (1:1 ratio) to perform competition experiments. We hypothesized that wild-type cells outcompete the BFP+ cells if DSBs induce p53/p21-mediated cellular senescence (Fig. 5D).
spCas9 transfection led to a significant GFP reduction in all cell lines; however, no correlation between GFP knockout and the loss of BFP+ cells was observed (Fig. 5E). This suggested the absence of p21-induced senescence. Next, the responses to RNP-induced DSBs in purified MPB CD34+ HSPCs were measured. As expected, HSPCs upregulated p21 (Fig. 5F) upon incubation with the p53-stabilizing agents Nutlin, resulting in significant cell death after 16 h (Supplementary Fig. 5F). This indicates that HSPCs are responsive to p53 activation and react by upregulating p21. Next, we induced multiple DSB breaks using different RNP complexes. Knockout efficiency assayed after 4 days was 60% for CD33 and 50% for CD45 (Supplementary Fig. S5G, H).
The double knockouts resulted in a somewhat lower knockout efficiency of ∼20% (Supplementary Fig. S5I). In contrast to treatment with the p53-stabilizing agent Nutlin, both the single and double RNP complex nucleofected CD34+ cells did not show significant upregulation of p21 (Fig. 5G). This suggests that p53 is either not activated due to RNP-induced DSBs or that this activation remains too low to detect using p21 as a readout.
Gene edited HSPCs show no defects on differentiation, proliferation, or lineage commitment
spCas9 potentially hampers cell proliferation or differentiation of HSPCs as suggested previously.
27
To test this hypothesis, knockouts were generated in HSPCs and differentiated toward erythroid cell cultures (Fig. 6A
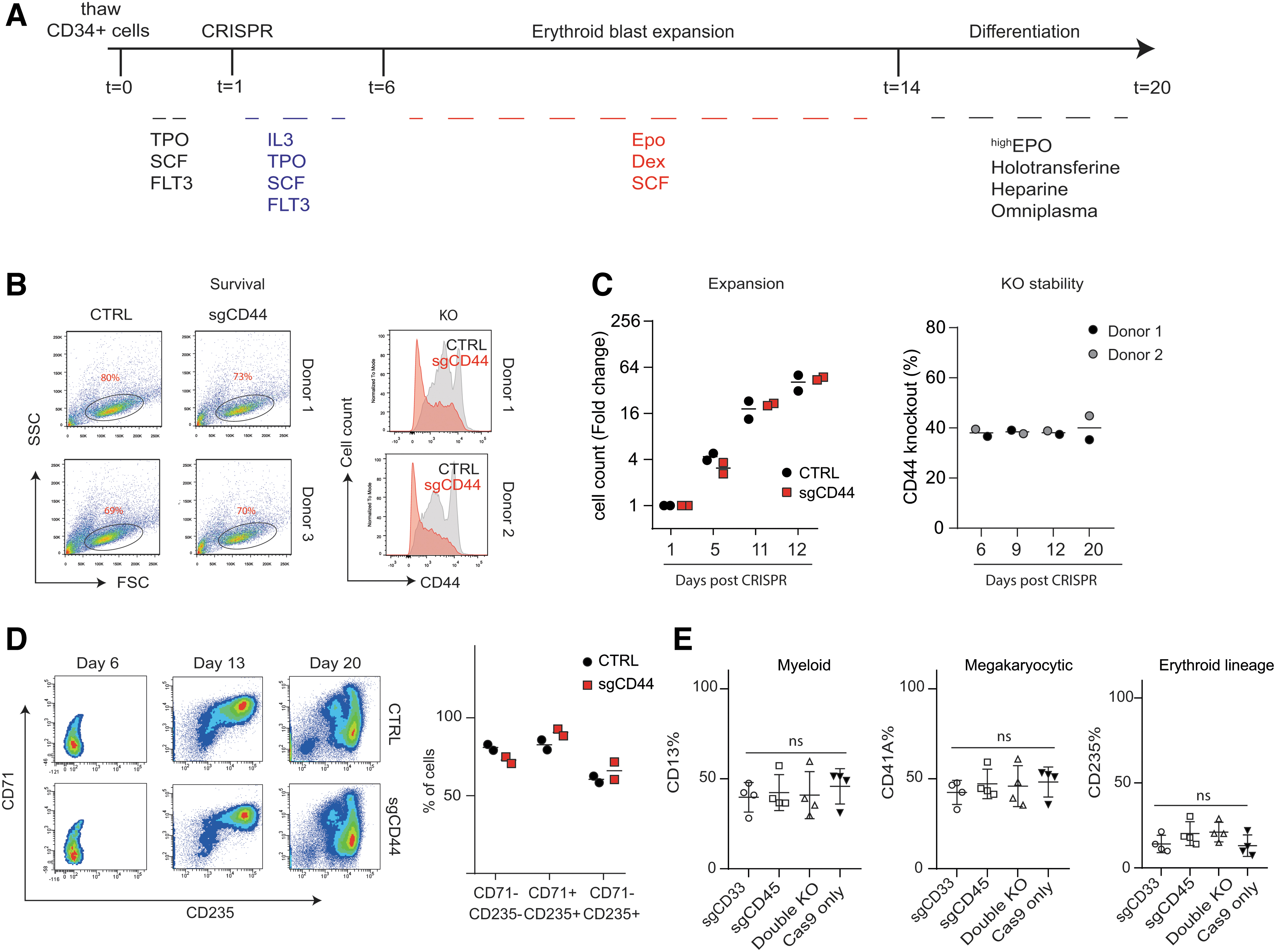
CRISPR-spCas9 has no effect on the differentiation or proliferation of HSPCs.
However, the frequency of CD13+ myeloid cells, CD235+ erythroid cells, and CD41+ megakaryoid/HSPCs did not change between cells that were successfully genome edited or that were nucleofected with spCas9 only (Fig. 6E). Importantly, the knockout efficiency within differentiated lineage cells was not altered compared to day 4. This showed that nucleofection as well as DSB induced by RNP complexes do not interfere with the specification to specific myeloid lineages. In addition, the data confirm that knockout of CD33, CD45, or the combination does not intrinsically interfere with differentiation to myeloid lineage.
Discussion
CRISPR-spCas9 gene editing is a powerful tool to study gene functions in the hematopoietic system. The protocols described here show that an optimized RNP system can result in high gene editing efficiencies in K562, independent of gRNA selection. As the turn-around-time of RNP applications is only 2 h, it substantially outperforms previous developed plasmid gene editing strategies. 13
Our data shows that the 3xNLS-spCas9 protein was greatly superior compared with 1xNLS 44 and 6xNLS 16 in gene editing within K562 and HSPCs. Concomitantly, 3xNLS-spCas9 was previously shown to outperform 2xNLSspCas9. 17 Surprisingly, similar gene editing efficiencies for 1xNLS 44 and 6xNLS 16 were observed, where it was previously demonstrated that 6xNLS significantly outperforms 1xNLS and 2xNLS-SpCas9 proteins by local delivery in mouse brain. 16 We speculate that the performance of spCas9 proteins and distinct fused NLS sites might be cell type dependent.
In general, our results indicate that genome editing in primary human HSPCs is less efficient compared with K562 and other cell lines that we have tested. This might be caused by (1) the nucleofection buffer, which is likely different for K562 compared with primary cells, and (2) the intrinsic nature of the cells that is clearly distinct for HSPC cells (e.g., DNA repair machinery or nuclear transport of RNPs).
Although high indel rates were previously reported,14,17 we find that achieving high gene editing efficiencies is target specific. gRNA selection is of critical importance to generate indel formations >20% in HSPCs, and this study demonstrates that in silico prediction models are not accurate in prediction. Although we included several reference gRNAs that previously showed high gene editing efficiency, such as for CD45 and CD33, 40 we show that indel rates are generally around 30% for the gRNAs we identified. Possibly, the efficiency might be improved by using synthetic sgRNAs or XT-gRNAs with extra chemical modifications, instead of the synthetic chemically modified hybridized ALTR-cr gRNA that was used in our research.
We find that IVTsgRNA screening in K562 leads to the successful prediction of effective gRNAs. The system that we setup can be easily applied in a resource poor setting as screening of IVTsgRNA is cost efficient and does not rely on primary cells. The addition of guanine on position 21 and 22 to boost expression might change the cleavage efficacy of the gRNAs and is in contrast to previously reported. 6 Our data show a distinct preference for nucleotide composition on multiple positions compared with the Wang algorithm, although the number of gRNAs used was limited to draw strong conclusions upon this. A possible reason why some in silico prediction models are ineffective can be the use of various cell type and transfer methods that was used to formulate these models.
HSPCs may be more difficult to target due to their active p53 status, compared to K562. Increased p21 mRNA expression levels were previously observed in the first 24 h using spCas9 by making a single DSB in HSPCs on the Y-chromosome of male cells. 27 Because the p53 pathway is actively involved in the non-homologous end joining route, we hypothesize that HSPCs are possibly more efficient in repairing the DSBs through p53-induced DNA repair machinery and thereby leading to decreased genome editing efficiencies. In fact, this suggests that transient p53 pathway inhibition using GSEA5627 can possibly enhance genome editing activities. It was previously shown that transient p53 inhibition using GSE56 prevents a proliferation delay in genetically modified HSPCs using AAV6 and finally resulted in better engraftment, which was found initially delayed in AAV6 genetically edited cells. 27
In our study, no significant p21 protein upregulation was observed in long term and suggests that gene editing in HSPCs does not affect cellular homeostasis. This is further supported by our data as no defects were observed in lineage commitment, proliferation, and differentiation. Because lineage commitment is not affected, we believe that gene editing in HSPCs is widely applicable to study any gene in correlation to specific differentiated cell types in the hematopoietic system such as myeloid blasts, erythroid cell types, and megakaryocytes.
Our data support the clinical trials that are currently ongoing and where the first patients are treated and show reconstitution of gene edited cells. 50
Footnotes
Acknowledgments
We thank Eelke Brandsma for critically reviewing the article. We also thank Niels Geijsen, Patrick Celi, Scott Wolfe, and Daniel Bauer for discussing the protein purification setup.
Authors' Contributions
H.J.M.P.V., C.K., A.M.K., L.R., S.A.M., and G.M. designed and performed the experiments. C.V. provided the CD34+ HSPCs. Concepts were formulated by H.J.M.P.V., E.A., and C.V. H.J.M.P.V., C.V., and E.A. wrote the initial article, and C.K., A.M.K., L.R., S.A.M., and G.M. made improvements.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by ZONMW game changer (116004203).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.