
Abstract
The introduction of small unmarked edits to the genome of insects is essential to study the molecular underpinnings of important biological traits, such as resistance to insecticides and genetic control strategies. Advances in CRISPR genome engineering have made this possible, but prohibitively laborious for most laboratories due to low rates of editing and the lack of a selectable marker. To facilitate the generation and isolation of precise marker-less edits we have developed a two-step method based on CRISPR-mediated cassette exchange (CriMCE) of a marked placeholder for a variant of interest. This strategy can be used to introduce a wider range of potential edits compared with previous approaches while consolidating the workflow. We present proof-of-principle that CriMCE is a powerful tool by engineering three single nucleotide polymorphism variants into the genome of Anopheles gambiae, with 5–41 × higher rates of editing than homology-directed repair or prime editing.
Introduction
Small genetic changes, such as single nucleotide polymorphisms (SNPs), can give rise to prominent phenotypes. For example, they are responsible for most genetic diseases in humans, 1 important agronomic traits in plants, 2 and insecticide resistance in insect vectors of disease. 3
To study their molecular underpinning, it is essential to engineer small precise edits such as these in the laboratory, 4 while excluding any transformation markers or gene editing debris that could interfere with the observed phenotype. The introduction of such marker-less edits has been facilitated by the discovery and expansion of clustered regularly interspersed short palindromic repeats (CRISPR) technologies.
In its most common form, CRISPR genome editing comprises a Cas endonuclease, able to catalyze a DNA double-stranded break; and a guide RNA (gRNA) that directs the Cas protein to its target sequence. 5 Simple and complex edits can be introduced with precision at a CRISPR-induced break by presenting a modified DNA template for homology directed repair (HDR). Recently developed base editing and prime editing methods are less versatile but work independently of the HDR pathway and can raise the efficiency of editing in species where HDR is naturally low.6–10
Base editing can induce transition point mutations through a Cas-deaminase fusion, whereas prime editing can introduce any point mutation or small indel by employing a Cas-reverse transcriptase fusion and a prime editing gRNA (pegRNA) that functions as a template for repair. 11 Neither have been widely tested in insects; however, initial trials in Drosophila suggest that prime editing is no more efficient than HDR, 12 whereas base editing is effective but inherently imprecise. 13
In insects, independent of the chosen technology, engineering small marker-less edits remains inefficient, with transformation rates rarely exceeding 5%.12,14,15 The lack of a molecular marker further hinders the process of identifying and isolating rare transformants, which becomes prohibitively laborious, relying on large numbers of single crosses and molecular identification of variants. Although there has been an expansion in the methods to engineer marker-less edits, this has not been met with a similar level of expansion in methods to isolate rare transformants.
We devised a two-step method to generate and facilitate the detection and isolation of precise marker-less edits, based on CRISPR-mediated cassette exchange (CriMCE) of a marked placeholder for a variant of interest (Fig. 1A). CriMCE relies on the visual detection of an edit, through the loss of a marker (Fig. 1A), which serves to enrich the pool of molecularly queried individuals for rare transformants, to reduce the labor and time required to isolate them (Fig. 1C).
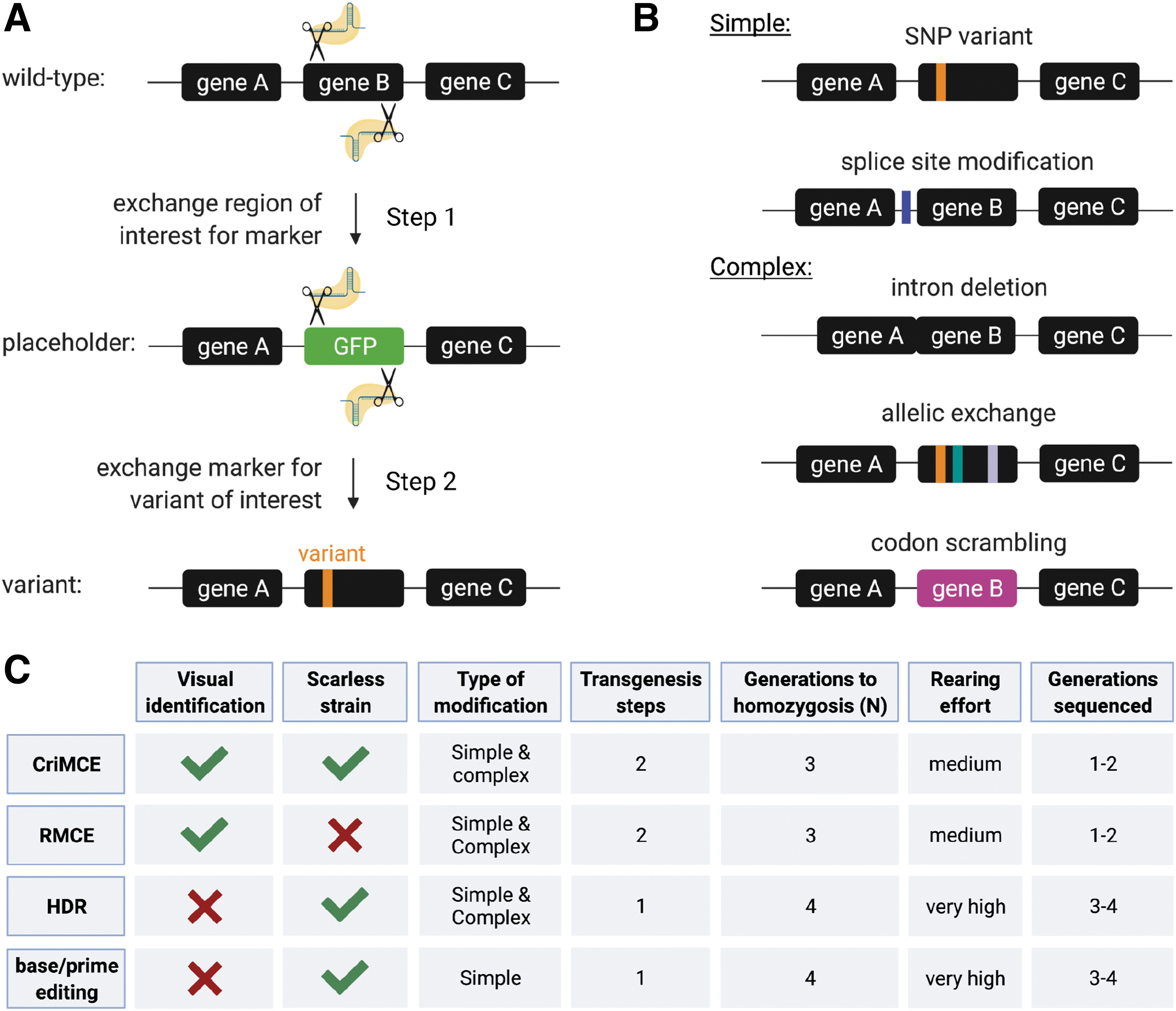
CriMCE is a two-step method for engineering the detection and isolation of marker-less edits through CRISPR-mediated HDR.
We demonstrate the value of CriMCE by deliberately introducing three SNP variants into the genome of the malaria mosquito, Anopheles gambiae, at the target site of a synthetic gene drive in the doublesex gene.16,17 Gene drives are engineered selfish genetic elements that show promise in controlling disease vector populations,16,18–20 but are susceptible to resistant mutations arising at the gene drive target site, in the form of SNPs or small indels.21–23 For vector control strategies, including insecticides and gene drive, it is becoming increasingly important to anticipate the emergence of resistance and pre-emptively design contingency plans. The SNP variant strains generated in this study will be useful in studying the potential for resistance to gene drives targeting a highly conserved site on doublesex and will inform implementation strategies.
We show that CriMCE is more efficient than methods previously employed to introduce small unmarked edits,12,14,15 while retaining versatility that would allow the engineering of more complex modifications as well (Fig. 1B).
Materials and Methods
Molecular cloning of CRISPR plasmids
We used Golden Gate cloning to insert a dual gRNA expression cassette into the p174 master vector, 16 to generate CRISPR vectors p174102 and p17404 needed to catalyze genomic cleavage for the insertion of a placeholder cassette and the variant of interest, respectively.
We first amplified a gRNA scaffold-U6 terminator-U6 promoter sequence, from plasmid p131 using primers containing BsaI sites (underlined), and gRNA sequences (capitals): BsaI-T1-U6-F (gag
Molecular cloning of placeholder donor plasmid
A 3xP3::GFP::SV40 marker cassette was amplified from plasmid pK101, 16 using primers SgsI-3xP3-F (GGCGCGCCCCACAATGGTTAATTCGAGC) and SgsI-SV40-R (GGCGCGCCAAGATACATTGATGAGTTTGGAC). Genomic DNA regions ∼1.8 kb upstream and downstream of the doublesex intron 4-exon 5 splice junction were amplified using primer pairs: 4050-KI-Gib1 (GCTCGAATTAACCATTGTGGACCGGTCTTGTGTTTAGCAGGCAGGGGA) with 4050-KI-Gib31 (TCCAAACTCATCAATGTATCTTGGCGCGCCATAAATGAATGGAAAGGTAAGGC), and 4050-KI-Gib32 (GAGCTCGAATTAACCATTGTGGGGCGCGCCGTATCTTTGTATGTGGGTGTGTG) with 4050-KI-Gib4 (TCCACCTCACCCATGGGACCCACGCGTGGTGCGGGTCACCGAGATGTTC), to make up the right and left homology arms, respectively, of the donor plasmid.
To generate the placeholder donor plasmid pHolder-dsx the three PCR products were combined with a digested vector backbone containing a 3xP3::DsRed::SV40 marker cassette in a four-fragment Gibson assembly, so that the doublesex (dsx) homology arms flank the green fluorescent protein (GFP) placeholder cassette.
Molecular cloning of variant donor plasmids
An intermediate plasmid (pVar-dsx) was Gibson assembled to contain the same vector backbone and homology arms as for pHolder, and a sequence containing BsaI cloning sites, flanking the region of interest of an otherwise intact exon 5 (Supp. Fig. 1A–C).
This allowed the Golden Gate cloning of annealed oligos containing three different doublesex exon 5 variants: a G → A SNP (GTTTAACACAGGTCAAGC
Embryo microinjections
An. gambiae G3 strain mosquitoes were reared at 26°C ± 2°C and 65% ± 10% relative humidity and blood-fed on cow blood using Hemotek membrane feeders. 18 Microinjections were performed on freshly laid embryos as previously described. 24 Each microinjected plasmid was present in solution at 300 ng/μL.
To generate the placeholder strain, wild-type embryos were microinjected with the p174102 CRISPR plasmid and pHolder donor plasmid (Supp. Fig. 1D–E). In transformants, this caused the excision of the coding sequence (CDS) of the female-specific exon 5 of the doublesex gene and its replacement with a GFP marker cassette. All microinjection survivors (G0) were crossed to wild-type mosquitoes and positive transformants (G1) were identified through fluorescence microscopy, as GFP+.
To generate the SNP variant strains, placeholder homozygote males were crossed to placeholder heterozygote females, distinguished using the complex parametric object analyzer and sorter fluorescence-based larval sorter. 25 Their progeny was microinjected with the p174104 CRISPR plasmid and each of the variant donor plasmids (pVar-dsxGA, pVar-dsxCT, and pVar-dsxGT) (Supp. Fig. 1D–E). In successful transformants, this caused the CriMCE of the marked placeholder for the doublesex exon 5 variants.
Injected survivors (G0) were distinguished from noninjected survivors (G0), as they exhibited red fluorescence in their posterior, due to successful injection of the p174104 CRISPR plasmid, containing a DsRed cassette in its backbone, which acted as a co-injection marker (Supp. Fig. 1D–E). All injected survivors (G0) were crossed to wild-type and females were deposited to lay eggs individually. A decreased inheritance of the marked placeholder (GFP+) in G1 progeny indicated CriMCE of the placeholder for the variant sequence (Fig. 2).
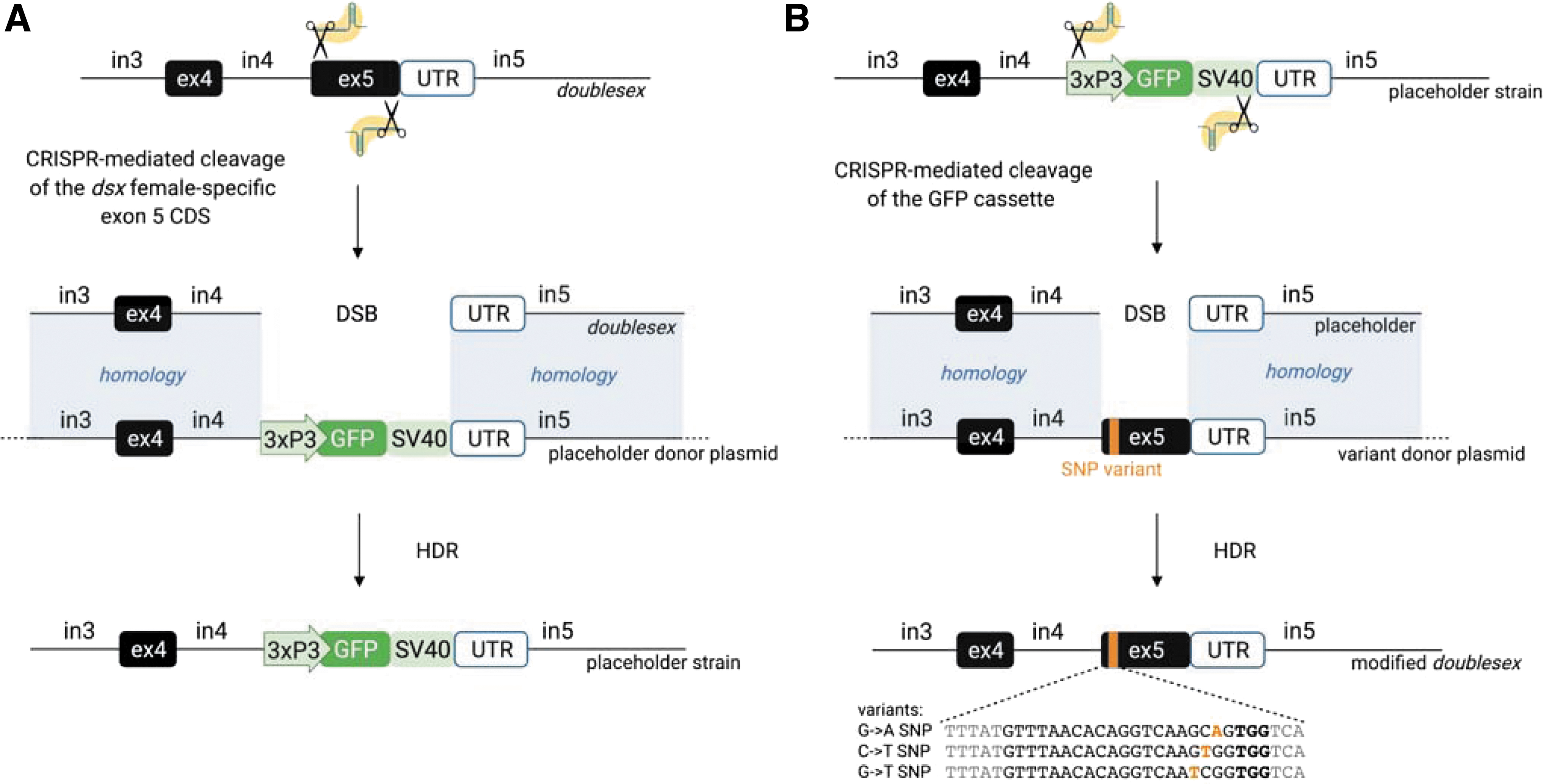
CriMCE relies on the generation of a marked placeholder strain, and the subsequent exchange of the placeholder for the variant of interest through CRISPR-mediated HDR.
Molecular genotyping
Genomic DNA was extracted from queried individuals after they gave offspring, in single samples, amplified using primers dsx-exon5-R4 (AACTTATCGGCATCAGTTGCG) and dsx-intron4-F1 (GTGAATTCCGTCAGCCAGCA) and sequenced using the dsx-exon5-R2 primer (TGAATTCGTTTCACCAAACACAC), to decipher their genotype.
Analysis
Figures were designed on Biorender (full license) and Adobe Illustrator and graphs were plotted and statistically analyzed on GraphPad Prism 9.
Results
We tested the efficiency of CriMCE and demonstrated proof of principle by using it to engineer and isolate mutations that potentially confer resistance to a gene drive, previously developed against the dsx gene in the malaria mosquito, An. gambiae. 16
First, we generated a placeholder strain by inserting a GFP cassette in place of the entire female-specific exon (exon 5) of dsx through CRISPR-mediated HDR (Fig. 2A). This strain was isolated based on GFP fluorescence, and displayed an intersex phenotype in homozygous females, consistent with the null mutation. 16
We then performed CriMCE of the placeholder for the marker-less SNP of interest (G→A, C→T, or G→T), by injecting placeholder homozygotes and heterozygotes with a plasmid expressing Cas9 and gRNAs targeted to the placeholder, and a template for repair encoding the variant of interest (Supp. Fig. 1D–E, 3B). To maximize the recovery of editing events, we selected only the fraction of injected mosquitoes that showed transient DsRed fluorescence as clear evidence of having taken up the CRISPR expression vector (Supp. Fig. 1D–E) and mated these to wild type (Fig. 3).
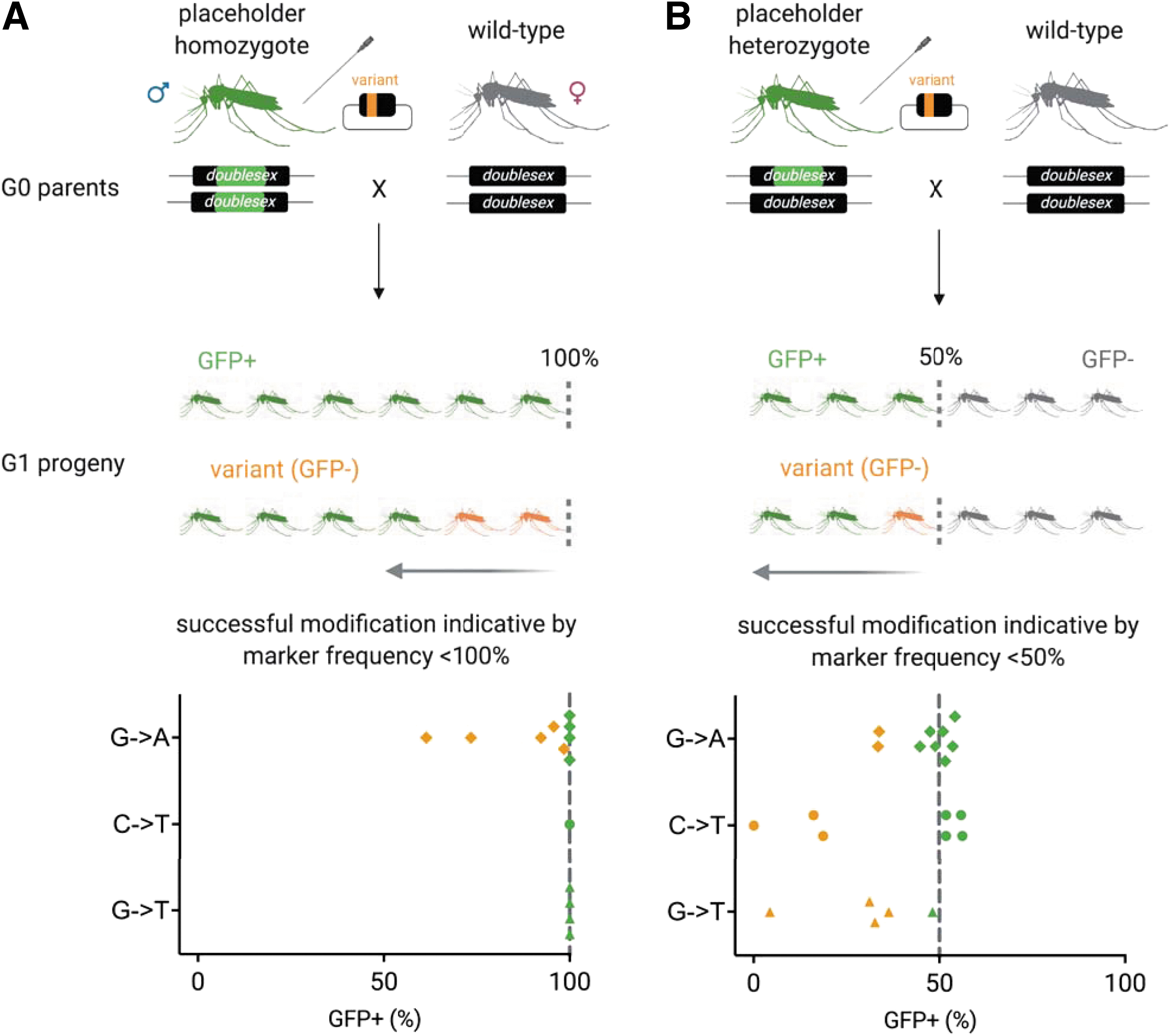
The introduction of a marker-less variant using CriMCE is evidenced by reduced rates of marker inheritance in the progeny of microinjected individuals of the placeholder strain. Marked placeholder male homozygotes
CriMCE-induced editing was evidenced by loss of GFP (<100% GFP inheritance) among the offspring of placeholder homozygotes, or by significant deviation below the Mendelian expectation of 50% GFP inheritance among the offspring of placeholder heterozygotes (Fig. 3). We saw rates of precise editing up to 39% for the G → A SNP (evidenced by 61% GFP inheritance in the offspring of placeholder homozygotes) (Fig. 3A), up to 100% for the C → T SNP, and up to 92% for the G → T SNP variant (evidenced by 0% and 4% GFP inheritance in the offspring of placeholder heterozygotes, respectively) (Fig. 3B). Incorporation of the SNPs of interest was confirmed by Sanger sequencing (Supp. Fig. 2). Notably, we did not detect any end-joining (EJ) events (N = 55). Owing to the high rates of editing by CriMCE, G1 transformants that showed low levels of GFP inheritance can be immediately crossed to the placeholder strain that will act as a balancer, for rapid characterization of each marker-less edit.
In two G1 clutches with altered GFP inheritance we also detected variant donor plasmid integration, evidenced by DsRed at 2% and 18% among GFP negatives (with a median of 0% taken across all modified clutches) (Supp. Fig. 1). These were not considered as true transformants in our analysis (Table 1 and Fig. 4).

Comparison of CriMCE with different transgenesis methods for the introduction of small precise marker-less edits. Welch's t-test p-values of statistical comparisons between CriMCE and prime editing are shown on top of each graph. HDR could not be statistically compared due to its small sample size.
Comparison of CRISPR-mediated cassette exchange with different transgenesis methods for the introduction of small precise marker-less edits or marked transgenes
Efficiency of each method is measured through the G1 transformant to G0 injected survivor ratio and the % of G1 transformants isolated from screened G1 progeny.
Only 18 out of 59 G0 injected survivors were kept and crossed to obtain G1 transgenics, due to COVID-19 restrictions in April 2020.
Identified visually.
The number of transformants is equal to the number of individuals lacking a fluorescent marker in the progeny of placeholder homozygotes. The number of transformant in the progeny of placeholder heterozygotes was estimated using this formula: (Total G1)/2 − GFP+ − DsRed+.
In most studies, G0 injected survivors are not being distinguished from noninjected survivors through transient expression of a fluorescent marker. The Kistler et al, 15 Gantz et al, 23 Hammond et al, 28 Adolfi et al, 20 and Ang et al 30 studies did not use such a method to distinguish injected survivors, or used all injected survivors (whether or not they showed signs of injection) to obtain transgenics.
Identified through sequencing.
Showing the set of injections with greater success for each method of prime editing: (a) using pegRNA expressed from a plasmid to provide cleavage and a template for repair, (b) using plasmid pegRNA together with an sgRNA to provide cleavage, (c) injecting a synthetic pegRNA straight away.
Note that the transgene integrated by HDR in the Gantz et al 23 study was significantly larger in size compared with all other studies, which could have reduced efficiency of integration.
The number of G0 founder pools that gave G1 transformants out of total G0 survivor pools is shown.
CriMCE, CRISPR-mediated cassette exchange; GFP, green fluorescent protein; HDR, homology directed repair; pegRNA, prime editing gRNA; RFP; RMCE, recombinase-mediated cassette exchange.
To compare our method with previously developed strategies employing HDR and prime editing to introduce and isolate marker-less edits,12,14,15 we calculated three measures of transformation efficiency: the percentage of G0 founders that gave G1 transformants, the G1 transformant to G0 injected survivor ratio, and the G1 transformant percentage out of all G1 screened (Table 1). If the G1 transformant to G0 injected survivor ratio is high, then a high number of transformants can be obtained from a smaller number of injected survivors; while having a high percentage of G1 transformants out of total G1 screened, implies a reduced requirement for screening, whether this is done visually, as in this study (less laborious), or by PCR and sequencing analysis, as in previous studies (more laborious). As a reference, we also show the efficiency of locus-specific marked transgene insertion through recombinase-mediated cassette exchange (RMCE) and HDR (Table 1).
In total, we detected visible editing in the progeny of 7/18 (38.9%) G0 microinjected individuals with the G → A construct, 3/8 (37.5%) G0 microinjected individuals with the C → T construct, and 4/9 (44.4%) G0 microinjected individuals with the G → T construct (Fig. 3 and Table 1).
CriMCE offers a marked improvement in transformation efficiency when compared with other approaches employed to introduce marker-less edits (Fig. 4). Specifically, CriMCE shows a mean G1 transformant to G0 injected survivor ratio of 5.76 (±2.37 SD), compared with 0.14 (±0.10 SD) for direct HDR (Welch's t-test p = 0.031) and 1.06 (±0.83 SD) for prime editing; and a mean G1 transformant per G1 screened percentage of 10.5% (±6.0% SD), compared with 1.0% (±0.5% SD) for direct HDR and 1.4% (±1.1% SD) for prime editing (Welch's t-test p = 0.058) (Fig. 4).
Discussion
To address the difficulty in engineering and isolating marker-less edits in insects, we have developed a strategy based on CriMCE of a marked placeholder for a variant of interest, allowing visual detection of transformation.
Unlike other two-step methods for marked cassette exchange or removal, such as RMCE and Cre-Lox recombination, CriMCE relies on HDR. This allows for comparatively high efficiency (when compared with RMCE) (Table 1), and uniquely traceless editing such that any phenotypic change can be attributed to the intended edit rather than ruminant attachment sites (Fig. 1). Co-conversion of a target locus together with a gene that produces a visual phenotype is another HDR-based strategy that has been used to improve isolation of marker-less edits. 26 This filters individuals showing CRISPR activity; however, it does not distinguish HDR events that incorporate the desired edit, from EJ events carrying unwanted indels. 26
Increasing the relative frequency of HDR over error-prone EJ repair remains difficult. Our strategy leverages loss of a marked placeholder (GFP+) to indicate precise editing by HDR. By targeting CRISPR to noncoding regions of the placeholder, undesirable EJ events are screened out as they are unlikely to affect GFP expression. Furthermore, we express Cas9 under the control of zpg regulatory elements that are spatiotemporally restricted to enhance HDR. 27 Indeed, no EJ mutations were detected in GFP-negative transformants. This focuses molecular identification by PCR and sequencing on individuals carrying the desired edit, therefore reducing the rearing effort required to enrich the frequency of marker-less variants (Supp. Fig. 3).
Somewhat surprisingly, rates of HDR-induced editing are relatively high when marked mutations are introduced (Table 1),23,28–30 but drop substantially when SNPs are directly inserted into a wild-type genomic locus, in Aedes aegypti and An. gambiae (Table 1 and Fig. 4).14,15 Using CriMCE in An. gambiae we achieved high rates of HDR editing consistent with those for marked edit insertion in An. gambiae and Drosophila melanogaster (Table 1 and Fig. 4).28,29 In both cases, repair templates differ significantly from their target regions: transgenes introduced through HDR do not resemble their genomic target, while in this study the wild-type target is replaced by a placeholder, which serves to differentiate it from the desired edit (Fig. 2).
Conversely, when direct HDR is used to induce small marker-less edits the repair template is almost identical to that of the wild-type target. It is still unclear why sequence dissimilarity between the exogenous repair template and its target should boost the efficiency of editing, but perhaps it functions to shift repair away from using the unmodified homologous chromosome as a template. Nonplasmid-based templates could also be used in a CriMCE strategy, such as single-stranded oligodeoxynucleotides that are simpler to produce and might further increase the rates of editing. 31
CriMCE might be less efficient in species with inherently low rates of HDR, such as Anopheles stephensi (Table 1),20,23 and alternatives not reliant on HDR, such as base and prime editing, 11 have not yet been tested in nonmodel insects. In these species, CriMCE can be optimized by injecting placeholder homozygotes, so that rare events are distinguished by visual inspection alone (Fig. 3A).
The CriMCE method can also mitigate against the risk of using previously untested and potentially inefficient gRNAs/pegRNAs that would otherwise expend undue effort on genetic crosses and molecular genotyping. Generating a marked placeholder before precise editing ensures that rare transgenesis using novel gRNA/pegRNAs is easily identifiable by a fluorescent marker. Previously tested guides can then be used to target the placeholder, inducing CriMCE. In this study, we validate the use of two gRNAs that target a universal placeholder, which is designed to function across insect species.
CriMCE is particularly powerful for experiments aimed at introducing a range of modifications to a single locus of interest, as a single placeholder strain can be exchanged for any number of variants. Indeed, a similar approach, based on exchange of a marked allele for engineering of kdr pyrethroid resistance mutations was employed in Drosophila, 32 and could be further extended to incorporate newly discovered insecticide-resistant SNPs. 33
Moreover, CriMCE allows for complex mutations that are not possible using prime editing since the entire region ablated by the placeholder can be replaced with a region bearing any number of desired edits. This strategy, which we term allelic exchange (Fig. 1C), could allow multiple linked SNPs to be introduced across a wide genetic locus. This would be useful in assessing how various resistant SNPs interact with each other to produce complex insecticide resistance phenotypes. 4 Other complex edits are also possible such as the introduction, modification or deletion of introns and splice site, or complete codon scrambling by which a CDS is modified without affecting the encoded amino acid sequence (Fig. 1C). The latter strategy could serve to engineer synthetic alleles that are resistant to gene drive elements as a mechanism for gene drive recall. 34
Finally, we describe how CriMCE can be used to target haploinsufficient genes, which by their nature, would be unable to tolerate a disruption from the placeholder, even if the desired edit is anticipated to be viable. In this case, integrating the placeholder within proximal intronic or neutral regions should permit editing (Supp. Fig. 4).
Conclusions
CriMCE is an efficient method to introduce and isolate precise and potentially complex marker-less edits by exchange of a visually marked intermediate. Our proof-of-principle experiments in An. gambiae suggest that CriMCE is 5–41 × more efficient than other strategies based on HDR or prime editing, while enabling an expanded range of potential edits and consolidating the workflow. In our experience, the use of a placeholder strain does not prolong isolation of the desired edit, and can be used as an important control or balancer in assessing its phenotype. We believe this strategy will be important in linking small genetic changes with a biologically relevant outcome across a range of insect species, with particular applications in the study of resistance to insecticides and gene drive technologies.
Footnotes
Authors' Contributions
The idea was conceived by I.M., A.C., and A.M.H. The experiments were designed by I.M. with input from T.N. and A.M.H. The experiments, data visualization, and analysis were performed by I.M. The original draft was written by I.M. and edited by A.M.H. The article was reviewed by all authors.
Acknowledgments
We thank Louise Marston, Matthew Gribble, and Molly McGrath for their help toward generating and maintaining transgenic strains.
Author Disclosure Statement
A.C. and A.M.H. are founders of Biocentis, Ltd. A.C., T.N., and A.M.H. have an equity interest in Biocentis, Ltd. I.M. declares no conflict of interest.
Funding Information
This study was supported by funding from the Medical Research Council (MRC) Doctoral Training Partnership (DTP) [1961745] and the Wellcome Trust [213694/Z/18/Z].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.