
Abstract
Ribonuclease III (RNase III) and RNase III-like ribonucleases have a wide range of important functions and are found in all organisms, yet a simple and high-throughput in vivo method for measuring RNase III activity does not exist. Typical methods for measuring RNase III activity rely on in vitro RNA analysis or in vivo methods that are not suitable for high-throughput analysis. In this study, we describe our development of a deactivated Cas9 (dCas9)-based in vivo assay for RNase III activity that utilizes RNase III's cleavage of the 5′-untranslated region (UTR) of its own messenger RNA. The key molecule in the system is a hybrid guide RNA (gRNA) between the 5′-UTR of RNase III and gGFP, a gRNA that works with dCas9 to repress GFP expression. This fusion must be cleaved by RNase III for full GFP repression. Our system uses GFP fluorescence to report on Escherichia coli RNase III activity in culture and on an individual cell basis, making it effective for selecting individual cells through fluorescence-activated cell sorting. Homology between enzymes within the RNase III family suggests this assay might be adapted to measure the activity of other enzymes in the RNase III family such as human Dicer or Drosha.
Introduction
Ribonuclease III (RNase III) is a double-stranded RNA (dsRNA)-specific endonuclease that cleaves dsRNA and has many regulatory roles in the cell. 1 RNase III homodimerizes to create a catalytic valley that allows cleavage of dsRNA within two identical active sites, leaving each strand with a characteristic 5′-phosphorylated, 3′-hydroxyl two nucleotide overhang.2–5 The RNase III family consists of related enzymes from bacteria to humans. Despite a wide range of functions, members of the RNase III family maintain a canonical nine residue signature motif. 6 Some enzymes within the family are Escherichia coli RNase III, which is responsible for ribosomal RNA processing and post-transcriptional gene regulation, and the more complex Homo sapiens Drosha and Dicer enzymes, which are responsible for processing the small RNAs involved with RNA interference.7–10
E. coli RNase III is a global regulator that post-transcriptionally modifies rRNAs and many messenger RNAs (mRNAs) by cleaving double-stranded structures within their coding and noncoding regions.3,5,11,12 Encoded by the rnc gene, RNase III acts as a homodimer, with each monomer consisting of an N-terminal RNase III endonuclease domain and a C-terminal dsRNA-binding domain that is important for dsRNA recognition.4,13
By comparing gene expression in strains with different levels of RNase III expression, several groups have found that numerous genes were differentially regulated, including genes related to cell growth and metabolism.14–16 In some circumstances, RNase III cleavage activates gene expression, such as with the adhE gene encoding for alcohol dehydrogenase. A stem-loop structure in the adhE mRNA must be removed to release the ribosome-binding site. 17 In other mRNA, cleavage by RNase III downregulates expression of the gene as cleavage leaves the mRNA vulnerable to degradation.18–20 RNase III natively autoregulates its own expression in this fashion. Within the 5′-untranslated region (UTR) of the mRNA of the rnc gene (Fig. 1), a stem-loop structure, loop II, protects the mRNA from degradation.
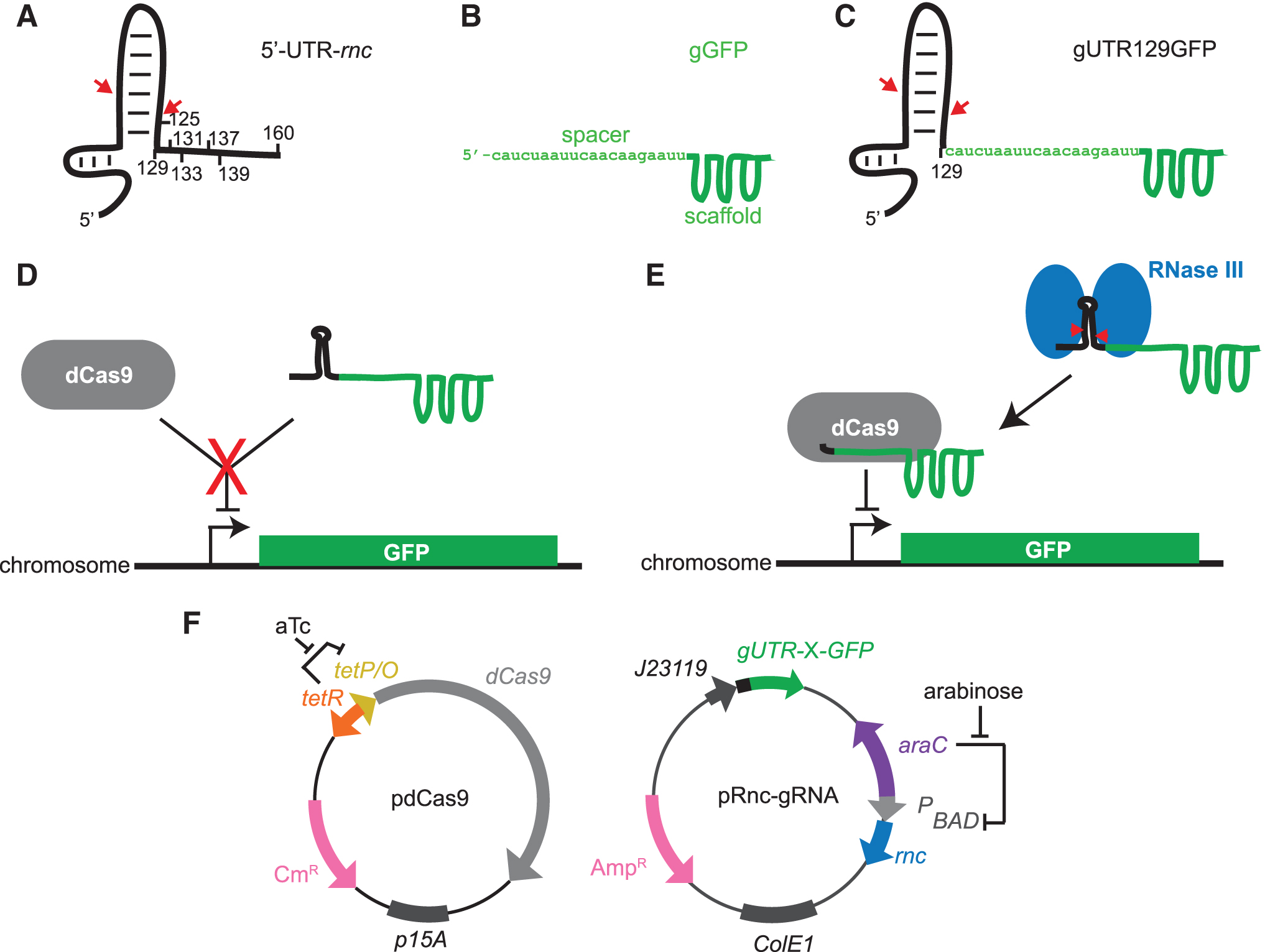
System for detection of RNase III in vivo.
In the case of overabundance of RNase III, expression is downregulated by negative feedback when RNase III cleaves loop II after positions 40 and 122 in its own mRNA, increasing the likelihood of degradation by RNase E.18,21–23 Despite its many important roles, RNase III activity is not essential for E. coli survival, but strains defective for RNase III activity grow slower than wild-type strains.24–26
The effect of mutations on RNase III is commonly studied by in vitro methods such as radiolabeled substrate cleavage assays, primer extension assays, and gel mobility-shift assays. Such methods allow researchers to quantitatively measure kinetic parameters for binding and cleavage but are low-throughput techniques that require purified protein. β-galactosidase (LacZ) has been used as a reporter of RNase III activity in vivo by fusing lacZ to the 5′-UTR of the phage lambda N gene 27 and 5′-UTR of rnc. 22 In the former assay, RNase III cleavage increases cellular β-galactosidase activity by releasing the Shine–Dalgarno sequence. In the latter assay, RNase III cleavage leads to an increased degradation of the lacZ mRNA. However, these assays have not been adapted to high-throughput analysis of RNase III mutants or to evaluation of RNase III activity on an individual cell basis, such as through use of flow cytometry. Such a method would be useful for analyzing libraries of mutant RNase III and could be used to select and identify interesting mutations to study.
We have developed a system that links RNase III activity in vivo to the expression of super-folder GFP (sfGFP) through the activity of deactivated Cas9 (dCas9). Cas9 is a site-selective endonuclease that complexes with a guide RNA (gRNA) and cleaves DNA at a site that is complementary to the gRNA. When the endonuclease domain is mutated to create a deactivated nuclease-null dCas9, it can act as a transcriptional repressor by blocking RNA polymerase.
Our system builds on the dCas9 system of Qi et al 28 in which a gRNA targeting a chromosomally integrated sfGFP represses sfGFP expression. Tang et al 29 demonstrated that this system could be extended to be ligand-dependent by fusing a ligand-responsive self-cleaving aptazyme to the gRNA. The aptazyme–gRNA remains nonfunctional because it cannot complex with dCas9 until the ligand causes aptazyme cleavage, releasing the gRNA to complex with dCas9 and repress GFP expression. In this study, we demonstrate an analogous system in which hybrid gRNAs are functionally compromised unless cleaved by RNase III. We demonstrate that fluorescence-activated cell sorting (FACS) can be used to selectively enrich for either active or inactive RNase III mutants using our system.
Materials and Methods
E. coli strains and growth conditions
Strain MG1693 (E. coli MG1655: K12 F− lambda− ilvG− rfb-50 rph-1 thyA715) and SK7622 (MG1693 Δrnc-38 KmR) were a gift from Sidney Kushner. The PH0919 strain was made by inserting a gBlock (Integrated DNA Technologies) based on the sequence from the strain used by Qi et al 28 containing sfGFP flanked by additional sequences (−182 bp from start codon and +177 from stop codon) into PH1105 (E. coli SK7622: PtauABCD::J23102 ΔtauD) genome at rbsAC locus using the method of Sabri et al. 30
Except where noted, all strains were grown in lysogeny broth (LB) media supplemented with 25 μg/mL kanamycin, 50 μg/mL ampicillin to maintain pRnc-gGFPX (where X is the nucleotide of rnc UTR that is fused to gGFP), and 50 μg/mL chloramphenicol to maintain pdCas9.
For analysis, strains were inoculated from colony or frozen glycerol stock in 5 mL of LB media as specified above and grown overnight at 37°C, shaking at 1.78 × g for 16 h. Inoculums were diluted 1/100 in 5 mL of fresh LB media and allowed to grow for 6 h, shaking at 250 rpm at 37°C. Experiments were performed in the presence and absence of arabinose (10–4 to 102 mM), and anhydrotetracycline (aTc) (2 nM), as indicated. All experiments were performed in cells transformed within 30 days because we noticed increasingly long lag times in frozen cells older than 30 days.
Vectors
pdCas9 (#44249) and pgRNA (#44251) were purchased from Addgene. The rnc gene (P0A7Y0) under control of the pBAD promoter and araC were amplified using Phusion High-Fidelity Polymerase (New England BioLabs) and inserted by blunt ligation between the 6 × Hig tag and pBAD reverse primer sequence on pgRNA. The rnc 5′-UTRs were fused by blunt ligation at nucleotide X along the 5′-UTR (where X = 125, 129, 131, 133, 137, 139, and 161) to the 5′ end of the gRNA spacer. For gUTR125GFP, the complimentary bases in stem loop II were altered to maintain Watson–Crick base pairing.
To make gRFP, the spacer targeting sfGFP was replaced by inverse PCR with the spacer targeting RFP. The RNase III E117K mutant was created by altering position 117 from GAA to AAA by overlap extension PCR. When used, the empty vector designates pRnc-gRNA:Δrnc lacking the rnc gene but maintaining the pBAD promoter and araC gene. pdCas9 ΔdCas9 was made by removing the dCas9 gene by inverse PCR using Phusion High-Fidelity polymerase. Vectors pRnc-gGFP129 (189537) and pRnc-gGFP161 (189536) are available at Addgene.
In vivo fluorescence reporting assay
Triplicates of 100 μL of each culture were added to a clear/black bottom 96-well microplate. Optical density at 600 nm (OD600) and GFP fluorescence at 485 nm excitation/525 nm emission was measured using a SpectraMax M3 microplate reader.
In vitro primer extension assay
The primer extension assay was performed essentially as described 31 ; the protocol is described in detail in this study. After 6 h of growth, 1 mL of 1 × 108 cells/mL were harvested and pelleted at 3000 g. The cell pellet was incubated in 200 μL of Tris-EDTA (TB) buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.4) (Gentrox) containing 1 mg/mL lysozyme at room temperature for 5 min, vortexing for 10 s every 90 s. A total of 680 μL of RLT (Qiagen) buffer with 1% 2-mercaptoethanol was added to the resuspended pellet followed by the addition of 490 μL of 100% ethanol. This lysate was used for RNA extraction with the RNeasy Mini Kit (Qiagen) according to the manufacturer's standard protocol for bacterial RNA purification.
The purified RNA was used to amplify complementary DNA (cDNA) from the gUTRGFP template with the 5′ Cy-5-labeled pWP252fluor primer (5′ ATAACGGACTAGCCTTA 3′) using the standard Superscript III First-Strand Synthesis System (Thermo Fisher) with 3 μg of total RNA and elongating at 52.5°C for 60 min. Next, undyed urea loading buffer (8 M urea, TE buffer) was added to cDNA and the DNA was denatured at 95°C for 3 min before loading in Novex 6% Tris-borate-EDTA (89 mM Tris base, 89 mM boric acid, 2 mM EDTA)-urea gel (Thermo Fisher). Samples were separated for 45 min at 180 V, 19 mA in TBE buffer (Quality Biological). The gel was visualized for Cy-5 fluorescence using a Typhoon 9410. Single-stranded DNA standards (ssDNA standards) were amplified from pRnc-gRNA DNA using Phusion High-Fidelity DNA Polymerase (New England BioLabs) with the pWP252fluor reverse primer and forward primers annealing to the expected 5′ termini of the cleaved and uncleaved 5′-UTR that would yield the desired size products.
PCR products were separated on a 4% agarose gel at 110V for 40 min and proper-sized products extracted using the QIAquick Gel Extraction Kit (Qiagen). Following denaturation in undyed loading buffer at 95°C for 3 min, DNA standards and cDNA both migrate as ssDNA in denaturing gels. Ethical review and approval were waived for this study because no human or animal subjects were involved in this study.
Flow cytometry
Six-hour cultures were diluted according to OD600 to a concentration of 1 × 106 cells/mL in cold phosphate-buffered saline (PBS). Samples were examined for GFP fluorescence using a BD Biosciences FACSCalibur flow cytometer on the green emission fluorescence channel 1 (FL-1). FlowJo 10.7 software was used to analyze raw data.
Fluorescence-activated cell sorting
Six-hour cultures were diluted according to OD600 to a concentration of 1 × 106 cells/mL in cold PBS. Strains carrying wild-type and E117K mutant RNase III were mixed in ratios of 80% wild-type/20% E117K and 20% wild-type/80% E117K. Pure cultures of each strain were used to set the gates for GFP fluorescence and forward scatter of wild-type and E117K populations using a Becton Coulter MoFlo Legacy cell sorter. Mixed samples were sorted according to these gates, collecting sorted populations from both gates for each mixture.
One hundred microliters of each presorted and sorted populations were plated on LB agar plates grown overnight in a standing incubator at 37°C. Random colonies from each plate were chosen for inoculation in 10 mL of LB media and grown overnight for 16 h, shaking at 250 rpm at 37°C. DNA was extracted from each culture using the standard MiniPrep (Qiagen) protocol. The rnc gene was PCR amplified from the plasmid with Phusion High-Fidelity polymerase and standard pBAD-Forward and pBAD-Reverse primers. Products were separated on a 2% agarose gel at 110V for 40 min and extracted using the QIAquick Gel Extraction Kit (Qiagen). Extracted products were Sanger sequenced using pBAD-Forward as a primer template by Genewiz. The fold enrichment (ɛ) was calculated from the initial (ri) and final (rf) ratio of target:nontarget cells using Equation 1.
Results and Discussion
Design of the system
We designed a reporter system for RNase III activity in vivo by adapting the fluorescent reporting system for dCas9 activity created by Qi et al. 28 In their system, the gRNA for GFP repression (gGFP) (Fig. 1B) directs dCas9 to repress expression of a chromosomally integrated Superfolder GFP gene (sfgfp). In our system, the gRNA responsible for GFP repression must be cleaved by RNase III for full repression. We fused a fragment of the 5′ UTR of the RNase III gene (rnc) (Fig. 1A), a native RNase III substrate, to the 5′ end of gGFP to create gUTRGFP (Fig. 1C). We designed gUTRGFP such that loop II in the rnc 5′-UTR sterically blocks the gUTRGFP from complexing with the dCas9, resulting in sfGFP expression (Fig. 1D). However, when RNase III is present, it cleaves the 5′-UTR from the gUTRGFP releasing the gGFP to complex with dCas9 and repress sfGFP expression (Fig. 1E). Our system consists of two plasmids, pdCas9 for expression of dCas928 and pRnc-gRNA for expression of gUTRGFP and RNase III (Fig. 1F).
The compatible plasmids are used in concert with PH0919, a Δrnc E. coli strain carrying the sfgfp gene on the chromosome. The pdCas9 plasmid has the dCas9 gene under control of the aTc-inducible promoter. Plasmid pRnc-gRNA encodes the gUTRGFP fusion under the control of the J23119 synthetic constitutive promoter and rnc under control of the pBAD promoter, which is induced by arabinose.
Design and optimization of rnc 5′-UTR length for the gRNA fusion
We first sought to identify which fragments of the 160-nucleotide long rnc 5′-UTR would interfere with GFP repression when fused to gGFP but still allow RNase III cleavage to release a functional gGFP. We sought to use stem-loop II to sterically interfere with gRNA function.
We designed seven gGFP fusions by fusing the 5′ end of gGFP to fragments of the rnc 5′-UTR (nucleotides 1-X; where X = 125, 129, 131, 133, 137, 139, and 161). Loop II in the 5′-UTR ends at residue 129. RNase III recognizes and natively cleaves within loop II of the 5′-UTR after nucleotides 40 and 12221–23 (Fig. 1A). In fusion gUTR129GFP, the 5′ end of the protospacer of gGFP is fused to the 3′ end of nucleotides 1–129 of the 5′-UTR (Fig. 1C). Fusions after nucleotides 131, 133, 137, and 139, moved the fusion site further away from loop II. Fusion gUTR161GFP used the full-length rnc 5′-UTR plus the first nucleotide of the coding region. We expected this fusion would be least likely to interfere with gRNA function because loop II would be furthest from the spacer. For fusion gUTR125GFP, we incorporated the first four bases of the gGFP into the stem of loop II and mutated the opposite strand to maintain the stem loop structure of loop II through Watson–Crick base pairing.
We tested the gUTRGFP fusions in the presence and absence of RNase III for the repression of the chromosomally integrated sfgfp. Initial experiments evaluating the effect of the inducers aTc (for dCas9 expression) and arabinose (for RNase III expression) suggested that fusions with shorter 5′-UTRs worked as designed, but the system worked better relying on the leaky expression of dCas9 in the absence of aTc (Fig. 2A, B).

Fluorescence of cells expressing 5′-UTR-rnc/gGFP fusions as a function of growth conditions and RNase III.
The presence of aTc caused increased GFP repression for all constructs even in the absence of RNase III, presumably from the increased levels of dCas9. In addition, we speculate that this added repression might result from shifting the equilibrium of the competition between the formation of stem loop II in the 5-UTR and the dCas9 complexing with the newly transcribed gUTRGFP. Another, nonmutually exclusive possibility is that elevated levels of dCas9 expression are toxic, and that toxicity affects GFP expression. Regardless of the mechanism, all subsequent experiments used leaky expression of dCas9 in the absence of aTc because we desired the largest difference in fluorescence between the presence and absence of RNase III.
We chose the tightly regulated pBAD promoter to tune the RNase III levels in the system because endogenous RNase III levels are only about 0.1% of total cellular protein, and we wanted to ensure expression is within the range that allows for normal cellular growth.15,16,23 To assess the effects of RNase III expression in our system, we analyzed how GFP expression varied with arabinose concentrations in the presence and absence of dCas9. We chose gUTR129GFP as a promising, representative gRNA fusion to examine in combination with the positive control of wild-type RNase III and the negative control of RNase III bearing the E117K mutation. E117K inactivates the catalytic activity of RNase III but does not inhibit binding to dsRNA.32,33
We found that our system functioned as designed at very low concentrations of arabinose and performed best without any arabinose (Supplementary Fig. S1). At ≤10 μM arabinose, only cells expressing wild-type RNase III were able to substantially repress GFP expression when dCas9 was expressed (Supplementary Fig. S1). High concentrations of arabinose had the undesired effect of repressing GFP expression in the absence of dCas9.
Furthermore, flow cytometric analysis of cells showed that high arabinose concentrations increased the forward scatter, suggesting that overexpression of RNase III causes an undesirable morphological change in the bacteria, potentially an increase in average cell size because of excess RNase III activity in the cell (Supplementary Fig. S2; Supplementary Table S1). As the system was working as expected, with RNase III expressed at desired levels even in the absence of the inducer arabinose, all subsequent experiments were performed in the absence of arabinose, except as noted.
We next evaluated all the 5′-UTR-gRNA fusion constructs in cultures lacking both aTc and arabinose (Fig. 2C). In the absence of RNase III, GFP expression depended on the 5-UTR-gRNA fusion site. Fusions closest to stem loop II (the 125 and 129 constructs) showed the least repression, consistent with our expectation that such fusions should sterically interfere with dCas9–gRNA complexation. Repression increased as the fusion site moved away from the stem loop, consistent with a reduction of steric interference of loop II in the formation of the dCas9:gGFP complex. In contrast, the presence of RNase III caused all fusions to repress expression to levels equivalent to that observed with an unmodified gGFP. Although the gUTR125GFP and gUTR129GFP fusions exhibit similar abilities in repressing GFP expression in an RNase III-dependent fashion, we chose to continue with gUTR129GFP because its unaltered loop II is a native substrate for RNase III.
Our experiments showed that the presence of RNase III repressed GFP expression, but they did not address how RNase III caused repression. The desired mechanism was that RNase III cleaves the gUTR129GFP, allowing the gRNA-containing product to interact productively with dCas9 to cause repression. However, another possibility was that RNase III repressed GFP expression by degrading the sfGFP mRNA. In the absence of dCas9, GFP is only marginally repressed by the addition of RNase III, suggesting that RNase III was not cleaving the sfGFP mRNA (Supplementary Fig. S3). However, we sought a more conclusive test in cells expressing dCas9.
We constructed a control gRNA (gRFP) that targeted the gene for the red fluorescent protein RFP, a gene absent from our cells. We evaluated the fluorescence of cells expressing gRFP, gUTR129GFP, and gUTR161GFP (a fusion construct that represses GFP expression even in the absence of RNase III). We evaluated GFP expression by measuring the fluorescence of the bacterial culture (Fig. 2D) and the median fluorescence of individual cells by flow cytometry (Fig. 2E).
Only gUTR129GFP could cause GFP repression with RNase III but not with the E117K mutant or the empty vector control. Cells with gRFP exhibited high GFP expression regardless of which RNase III was expressed, and gUTR161GFP caused repression regardless of which RNase III was expressed. Our system produced the desired RNase III-dependent fluorescent signal that was consistent with our proposed mechanism in which RNase III cleaves gUTR129GFP to activate it for complexation with dCas9 to repress GFP expression.
One curious result in the experiments using gRFP was that fluorescence/OD decreased when RNase III was expressed relative to cells lacking a functional RNase III (Fig. 2D), yet when the cells were analyzed by flow cytometry, cells with a functional RNase III had a roughly twofold higher fluorescence. We speculate that this difference results from RNase III's effects on cell size and morphology. In the absence of arabinose, expression of RNase III in PH0919 causes a twofold increase in forward scatter when the cells are analyzed by flow cytometry (Supplementary Table S1), which suggests that cells with a functional RNase III are larger. Larger cells would scatter more light, thus decreasing fluorescence per OD (Fig. 2D), but might have more fluorescence simply because they were larger (Fig. 2E).
RNase III cleaves the 5-UTR/gRNA fusions as expected
We next sought to confirm that the gUTR129GFP fusion was uncleaved in the absence of RNase III and cleaved when RNase III was expressed. We devised a primer extension assay to evaluate the length of RNA containing the gRNA end of the fusion (Fig. 3A). We used extracted mRNA from cultures as a template for primer extension using a 5′-Cy5-labeled primer specific to the nucleotides 50–66 within the dCas9 handle of the gRNA.
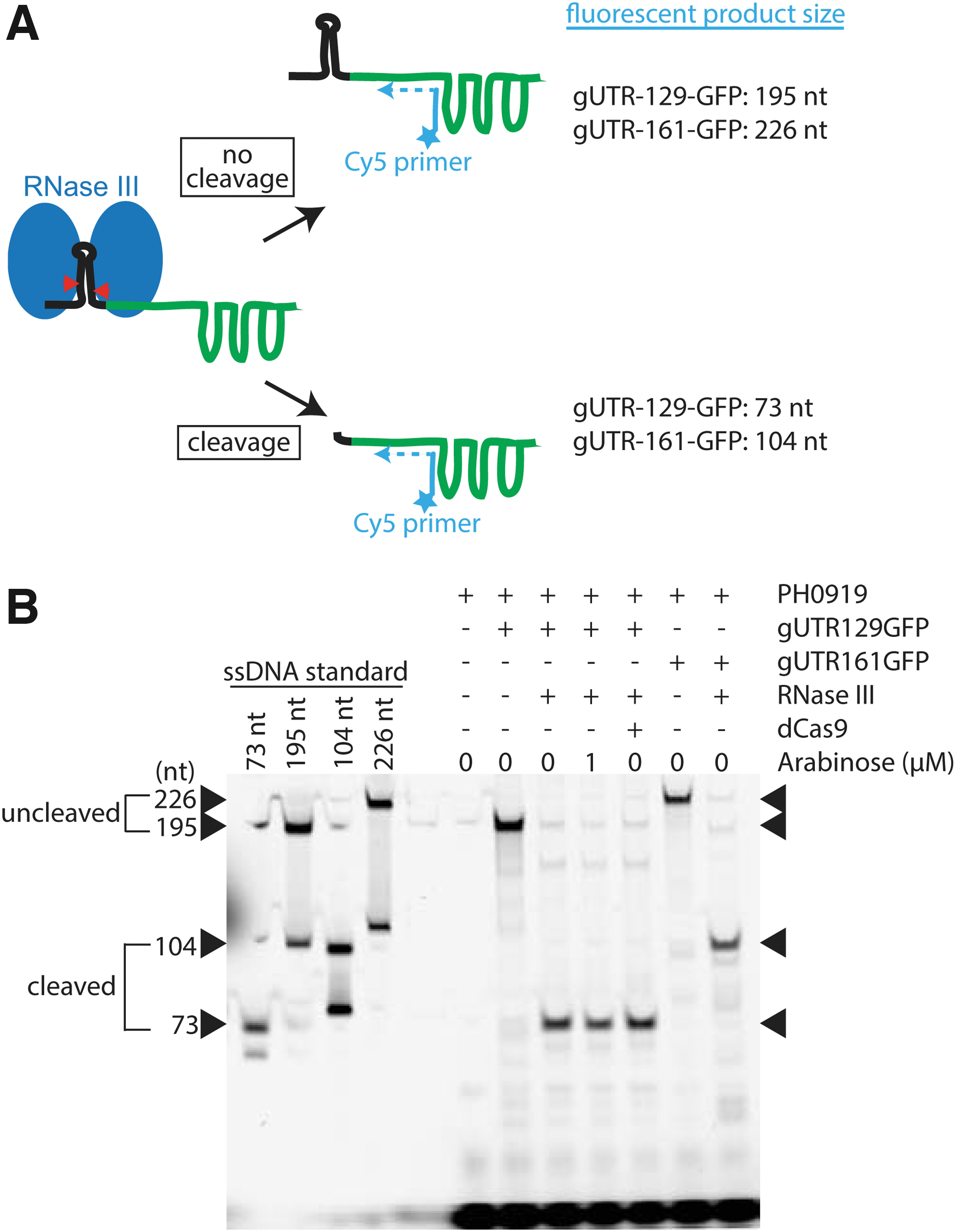
RNase III selectively cleaves gUTR129GFP and gUTR161GFP in vivo.
We first confirmed that RNase III expressed from pRnc-gRNA in the absence of arabinose-cleaved gUTR129GFP as effectively as chromosomally expressed RNase III and that RNase III with E117K does not cleave gUTR129GFP (Supplementary Fig. S4). In cells lacking RNase III or containing the E117K mutant, we observed bands at the expected length of 195 for uncleaved gUTR129GFP. In the presence of RNase III, however, the band shifted to 73, corresponding to the expected length of the cleaved gUTR129GFP. We next confirmed that an analogous RNase III-dependent change in size occurred when gUTR161GFP was expressed and that the presence of dCas9 does not prevent RNase III cleavage of gUTR129GFP (Fig. 3B).
The shift of the bands in the presence of RNase III to the expected length is consistent with RNase III cleaving the 5′-UTR in loop II. Given that RNase III cleaves both the gUTR129GFP and gUTR161GFP fusions, but only gUTR129GFP requires RNase III to repress GFP expression, we conclude that the 5′-UTR portion of gUTR129GFP (likely loop II) interferes with dCas9 binding of the gUTR129GFP fusion. Increasing the distance between loop II and the protospacer allows for a normal dCas9–gRNA interaction in the absence of loop II cleavage. This hypothesis is supported by the pattern of repression observed with the various 5′-UTR-gRNA constructs in Figure 2C.
FACS sorting can be used to enrich for active or inactive RNase III variants
Our observation by flow cytometry that cells expressing wild-type and E117K mutant RNase III have an ∼5-fold difference in fluorescence (Fig. 2E; Supplementary Fig. S4) suggested that FACS could be used to isolate active or inactive RNase III variants from a population of cells.
We tested this idea by enriching active RNase III from an excess of inactive RNase III and inactive RNase III from an excess of active RNase III. We grew cultures of cells expressing dCas9/gUTR129GFP and either wild-type RNase III or the E117K mutant. Once the cells reached exponential growth, we combined the cultures based on optical density to make mixed cultures of 80% wild-type/20%E117K and 20% wild-type/80%E117K. We performed FACS on these cultures using fluorescence and forward scatter to set the sorting gates to enrichment for the desired minor population in the mixture (Supplementary Fig. S5). We plated cells before and after sorting then sequenced the DNA of at least 20 individual colonies to determine the frequency of wild-type and the E117K mutant. In both cases, cells that were in the minority before the sort became the majority after the sort, and the enrichment was on the order of 100-fold (Table 1).
Fluorescence-Activated Cell Sorting Enrichment of Active and Inactive RNase III
Number of colonies containing the indicated variant as determined by DNA sequencing.
Calculated from Equation 1 using a 1:4 initial ratio of target:nontarget cells.
Calculated from Equation 1 using the observed initial ratio determined by DNA sequencing.
WT, wild-type.
Potential uses
Previously, more laborious in vitro procedures such as northern blotting or primer extension analysis would be needed to evaluate RNase III mutants' activity on their substrates. Our system allows us to easily characterize individual RNase III mutants as active or inactive in vivo based on the level of cell fluorescence. Moreover, we have found that intermediate levels of RNase III activity in the cell will produce intermediate levels of cell fluorescence (Weeks and Ostermeier, in preparation). In this unpublished work, we have used our system to characterize a comprehensive library of RNase III mutants by sorting the library by FACS into bins of high and low fluorescence and then using next-generation sequencing to identify the frequency of each mutation in each bin. From this mutation frequency data, we quantified the effect of the mutation on cellular RNase III activity on a continuous scale, as is typically done in deep mutational scanning studies. 34 Additionally, our general strategy could be used to study related ribonucleases with substrates that possess secondary structures.
What is required is fusion of a ribonuclease's substrate to the gGFP, such that it sterically blocks the gGFP's functional complexation with dCas9, but cleavage of the substrate releases a functional gRNA. Thus, our approach may find use for other bacterial ribonucleases and related human ribonucleases such as Drosha and Dicer. Achieving an equivalent system for ribonucleases other than E. coli RNase III will require identifying a useful substrate–gGFP fusion as well as a careful balancing of the expression of dCas9 and the ribonuclease, as was required in this work.
Footnotes
Acknowledgments
The authors thank Steven Kushner for strains MG1693 and SK7622, Hao Zhang for support with FACS, and Indra Mani Sharma and Sarah A. Woodson for assistance with the primer extension assay.
Authors' Contributions
M.O. conceived the study. P.H., R.W., and M.O. designed experiments. P.H. and R.W. performed experiments, data analysis, and statistical analysis. R.W. and M.O. wrote the article with input from P.H. All authors approved the article for publication.
Author Disclosure Statement
The authors declare no competing conflicts of interest.
Funding Information
This research was supported by the National Science Foundation Grant MCB-1817646 and Pebble Labs (Los Alamos, NM).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.