
Abstract
Advances in molecular medicine have placed nucleic acid detection methods at the center of an increasing number of clinical applications. Polymerase chain reaction (PCR)-based diagnostics have been widely adopted for their versatility, specificity, and sensitivity. However, recently reported clustered regularly interspaced short palindromic repeats-based methods have demonstrated equivalent to superior performance, with increased portability and reduced processing time and cost. In this study, we applied Specific High-Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK) technology to the detection of oncogenic rearrangements. We implemented SHERLOCK for the detection of BCR::ABL1 mRNA, a hallmark of chronic myeloid leukemia (CML), and EGFR DNA oncogenic alleles, frequently detected in glioblastoma and non-small cell lung cancer (NSCLC). SHERLOCK enabled rapid, sensitive, and variant-specific detection of BCR::ABL1 and EGFR alterations. Compared with the gold-standard PCR-based methods currently used in clinic, SHERLOCK achieved equivalent to greater sensitivity, suggesting it could be a new tool in CML and NSCLC, to detect low level of molecular residual disease.
Introduction
Cancer patient care is rapidly evolving toward personalized medicine. Systematic profiling of somatic mutations in a wide range of tumor types has led to the identification of specific molecular subtypes. This may guide clinical decision and treatment choice. 1 Nucleic acid biomarkers are now frequently used not only to help diagnosis and evaluate the prognosis but also for minimal residual disease monitoring and relapse assessment. Consequently, detection of nucleic acid biomarkers has been placed at the center of an increasing number of clinical and diagnostic applications. 2 Chronic myeloid leukemia (CML), non-small cell lung cancer (NSCLC), and glioblastoma (GBM) reflect well tumor molecular subtyping for the development of targeted therapies and adapted treatment. CML is a rare hematologic cancer that results from an acquired t(9;22)(q34;q11) reciprocal translocation generating the BCR::ABL1 fusion oncogene.
The introduction of targeted therapies based on tyrosine kinase inhibitor (TKI) revolutionized CML treatment by achieving outstanding effectiveness leading to unprecedented increased life expectancy of patients close to the age-matched healthy population. 3 In the same manner, the identification of EGFR mutations in NSCLC tumors has introduced the selective use of TKI and increased progression-free survival and overall survival of patients. Specific EGFR rearrangements, larger than those observed in NSCLC, are also frequent in GBM. 4 This cancer is the most common malignant brain tumor in adults and one of the most lethal cancer type. 5 Therefore, targeted therapies elicit great hope to treat GBM. In all three cases, accurate identification and follow-up of respective biomarkers allows the monitoring of cancer remission or relapse6,7 and helps patient management. 8
Polymerase chain reaction (PCR)-based detection of biomarkers has been widely adopted for their versatility, specificity, and sensitivity. Real-time quantitative PCR (RT-qPCR) and digital droplet PCR (ddPCR) are used for the detection and quantification of known biomarkers. RT-qPCR has emerged as the assay of choice for monitoring CML evolution and the treatment response by assessing BCR::ABL1 transcript levels.9,10 Similarly, TKI treatment response of NSCLC patients is routinely monitored with ddPCR by quantifying the levels of mutated EGFR alleles in circulating tumor DNA (ctDNA) extracted from liquid biopsy samples.
The assay sensitivity is a key parameter to perform optimal liquid biopsy analysis and track cancer evolution. Ideally, negative test results should be associated to complete disappearance of the biomarker rather than lack of sensitivity of the detection method. However, the first STop IManitib trial reported frequent molecular relapse (61%) after imatinib cease in CML patients who had negative BCR::ABL1 RT-qPCR for at least 2 years.6,7 This suggests that RT-qPCR fails to detect low level of minimal residual disease. Similarly, detection of EGFR mutations in ctDNA using ddPCR has shown promising results, outperforming standard qPCR. 11 Despite their sensitivity, ddPCR, and to a lesser extent RT-qPCR, rely on costly equipment, trained personnel, and expensive reagents. However, one of the major challenges of the coming decades is to ensure equitable access to care, including molecular medicine. This requires the development of inexpensive methods, using routine laboratory equipment. 12
The recently reported clustered regularly interspaced short palindromic repeats (CRISPR)-based diagnostics carries the potential to enable cost-effective and sensitive detection of BCR::ABL1 and EGFR rearrangements, offering a valuable alternative to gold-standard RT-qPCR and ddPCR. 13 The use of the CRISPR-Cas system has enabled the sensitive and fast detection of nucleic-acid-based biomarkers opening exciting opportunities in nucleic-acid based diagnosis. Among the Cas nucleases, the class 2 type VI Cas13a ribonuclease is capable of specific detection and cis-cleavage activity of single-stranded RNA (ssRNA) target.14,15 Cleavage of the RNA target activates the Cas13 trans-cleavage activity leading to RNA nonspecific targeting.
Based on the specific properties of the Cas13a ribonuclease from Leptotrichia wadei (LwCas13a), Gootenberg et al. developed the Specific High-Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK).16,17 SHERLOCK is a two-step process (Supplementary Fig. S1a, b). First, DNA or RNA of interest is amplified from trace amounts using isothermal amplification followed by in vitro transcription. Then, amplified RNA products and a short ssRNA reporter, including a fluorophore-quencher pairs, are submitted to LwCas13a detection and cleavage.
Cas13-mediated specific target cleavage activates its collateral trans-cleavage activity, leading to ssRNA reporter degradation, spatial separation of the fluorophore and the quencher, and thus to fluorescence emission. SHERLOCK enables rapid, sensitive (10−18–10−21 M)16,18 and, field-deployable detection of DNA and RNA and have already been applied to detect the presence of exogenous sequence (virus and bacteria) in the human genome,16,18,19 including Sars-Cov-2,20,21 from various biological samples, but also mRNA, 22 and miRNA, 23 endogenous markers.
In this study, SHERLOCK was implemented for the detection of oncogenic rearrangements. We first adapted SHERLOCK to the detection of BCR::ABL1 fusion transcripts and to two distinct EGFR DNA oncogenic alleles (EGFRvIII and EGFRdel19). Then, we compared the sensitivity of SHERLOCK with the gold-standard detection methods used for the detection of BCR::ABL1 oncogenic fusion transcripts and specific EGFR oncogenic rearrangements. Finally, we evaluated the ability of SHERLOCK to detect EGFRdel19 mutations in NSCLC patient liquid biopsy samples. Collectively, our findings demonstrated that SHERLOCK represents a valuable option to current methods for the detection of oncogenic rearrangements.
Materials and Methods
Cell culture and cell sorting
K-562, KCL-22, and HL-60 cell lines (ATCC®, Manassas, VA) were maintained in Roswell Park Memorial Institute medium 1640 supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cell lines were cultured at 37°C, 5% CO2 in a humidified chamber. Cell dilutions were performed by sorting a defined number of KCL-22 or K-562 (target cells) to 10 million HL-60 (nontarget cells) with BD FACS Aria®. KCL-22 and K-562 were gated on morphology.
RNA extraction and cDNA synthesis
RNA from K-562, KCL-22, and HL-60 were extracted with phenol/chloroform. In brief, cell pellets were resuspended in 1 mL TRIzol® (Invitrogen, MA) and mechanically disrupted for 10 min. RNA was separated using chloroform:isoamyl alcohol 24:1 (Sigma-Aldrich, MO), precipitated with isopropanol, washed with 70% ethanol, and resuspended in nuclease-free water. The input of 1.0 μg RNA was used to generate cDNAs with SuperScript™ VILO™ cDNA Synthesis Kit (Invitrogen) following the manufacturer's instructions.
BCR::ABL1 expression quantification in cell dilutions
The quantification of ABL1 and BCR::ABL1 cDNA copy number was performed by qPCR according to the Europe Against Cancer (EAC) protocol using ABL1 as an internal control gene. 24 Eighteen microliters of H2O were added to the 12 μL of cDNA and 5 μL of the diluted cDNA (for a total of 100 ng per replicate) were then amplified using the primers and probes of the EAC (Supplementary Table S1) and LC480 PROBES MASTER MIX® (Roche) on LC 480 Thermocycler® (Roche). Three replicates of BCR::ABL1 and two replicates of ABL1 were performed. ABL1 and BCR::ABL1 copy numbers are shown in Supplementary Table S4.
Patient samples and DNA extraction
EGFRvIII and EGFRdel19 DNA extracts were obtained from tumor patient samples and extracted using Maxwell RSC formalin-fixed paraffin-embedded (FFPE) DNA extraction kit® (Promega, WI). Initial assessment of EGFRvIII DNA breakpoints positions in intron 1 and intron 7 of the EGFR gene was obtained by next generation sequencing (NGS) using a Foundation One® (Boston, MA) large panel analysis. Similarly, initial assessment of the EGFRdel19 mutation (c.2236_2250del) was performed by NGS using in-house ampli-seq based panel on Ion S5® (Thermo Scientific) sequencing device at the somatic molecular biology department of Bordeaux University Hospital.
Wild-type DNA sample was extracted from healthy donor buffy coat using QIAamp DNA extraction kit® (Qiagen). Circulating DNA from plasma samples were obtained from peripheral blood of NSCLC patients with tumors harboring the EGFR exon 19 c.2236_2250del mutation (confirmed by NGS). DNA were extracted using Maxwell RSC ccFDNA plasma extraction kit® (Promega). All DNA samples were quantified by spectrophotometry using Nanodrop® One/One device (Thermo Fisher Scientific).
Serial DNA dilutions
To perform serial DNA dilutions, allele-specific EGFRmutated and EGFRwt primer pairs were designed to amplify EGFRmutated or EGFRwt, respectively (Supplementary Table S1). qPCR were performed using the Promega GoTaq® qPCR kit (Promega). The comparative Mean Normalized Expression method was used to quantify EGFRwt DNA extracted from normal white blood cells, and EGFRvIII or EGFRdel19 alleles from tumor DNA samples. Tumor and normal DNA were then mixed according to the qPCR quantification to obtain an initial 10 ng·μL−1 dilution with 50% of mutated copies and 50% of wild-type copies. In this mix, the number of copies of EGFRwt was quantified using an external DNA control allowing the calculation of the corresponding copy numbers of EGFRvIII and EGFRdel19 in the parental solution and the subsequent dilutions (Supplementary Table S4).
Amplification of BCR::ABL1 cDNA, EGFR gDNA, and ctDNA
e13a2-BCR::ABL1 and e14a2-BCR::ABL1 PCR products were generated by amplifying 8 μL of cDNA with e13a2- or e14a2-specific primers with the GoTaq G2 DNA Polymerase (Promega). EGFR PCR products were amplified with Phire Tissue Direct PCR Master Mix® (Thermo Fisher Scientific) using a 10 ng input of gDNA or ctDNA (1 μL concentrated at 10 ng·μL−1). Amplification primers are listed in Supplementary Table S1. An overhang including the T7 promoter was used to enable subsequent T7-mediated in vitro transcription of the PCR products (see Kellner et al.). 17 By default, 35 cycles of amplification were performed.
Real-time qPCR
Detection of e13a2, e14a2, and EGFRvIII was performed using the Promega GoTaq qPCR kit (Promega) and a CFX96 Touch Real-Time PCR Detection System (Bio-Rad), following manufacturer's instructions guidelines. Primers used are in Supplementary Table S1. Data were analyzed with CFX Maestro Software (Bio-Rad). Relative expression of BCR::ABL1 and EGFRvIII gene was first normalized to GUSB and GAPDH, respectively, and then represented as fold changes (2-ΔΔCt). Melting curves showed that primers amplified only the specific fragments.
Production of guide RNAs
Guide RNA were produced by T7-mediated in vitro transcription as described in Kellner et al. 17 In brief, oligonucleotides (PAGE Ultramer DNA oligos from Integrated DNA Technologies) were annealed and in vitro transcribed overnight with the HiScribe™ T7 Quick High Yield RNA Synthesis Kit (NEB, MA) following manufacturer's instructions and subsequently purified with Agencourt RNAClean XP beads (Beckman Coulter). Purification products were aliquoted and frozen at −80°C before use.
SHERLOCK detection
In vitro transcription of BCR::ABL1 and EGFR PCR products and Cas13-mediated detection of T7-produced RNA were performed simultaneously as described previously. 17 Detection mix included 16 mM HEPES, 7.2 mM MgCl2, 640 nM rNTP, 0.05 U·μL−1 T7 RNA polymerase, 1.6.10−3 U·μL−1 murine RNase inhibitor (NEB), 5 μg·μL−1 LwaCas13a (ordered from GenScript and stored at −80°C in 50 mM Tris-HCl, 600 mM NaCl, 5% Glycerol, 2 mM DTT, and pH 7.5), 400 pg·μL−1 crRNA, 100 nM fluorescent RNA reporter and 5 μL of PCR products. The total volume for one reaction was 25 μL.
Reagents stock concentrations, total quantities for one reaction and final mix concentrations are described in Supplementary Table S3. crRNA and RNA reporter sequences are described in Supplementary Table S2. Mix was prepared on ice to limit reaction initiation before fluorimeter plate reading. After the addition of PCR products, samples were quickly transferred to a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) and the fluorescence level was quantified every minute. Result analyses were performed using CFX Maestro™ software (BioRad).
Statistical analysis
Statistical significance was inferred when necessary. The Graph Pad Prism 6 software was used for statistical analysis. One-way Brown–Forsythe analysis of variance tests completed with unpaired t-test with Welch's correction were used. Exact p-values are shown or represented as **for p-value <0.01.
Ethic statement
The study was approved by the Ethics and Research Committee of Bordeaux university Hospital and conducted according to the ethical guidelines of the 1975 Declaration of Helsinki.
Results
SHERLOCK enables the specific and sensitive detection of BCR::ABL1
For more than 95% of CML patients, the breakpoint in BCR occurs in the major breakpoint cluster region leading to the e13a2 or e14a2 transcript and up to 10% of CML patients are positive for both variants (Supplementary Fig. S2a, b). 25 Unlike e13a2 isoform, the e14a2 transcript variant harbors BCR exon 14. We first designed two RNA guides targeting either the e13a2 or the e14a2 isoforms. Since ABL1 exon 2 is common to the e13a2 and e14a2 isoforms, the first 15 nucleotides of the two guides targeting this region are identical, potentially raising a specificity issue.
To evaluate the specific detection of each transcript variants, we performed SHERLOCK detection on RNA samples from KCL-22 and K-562 leukemic cell lines, expressing the e13a2 or the e14a2 isoforms, respectively (Fig. 1a). Highly specific detection was achieved when the RNA guide matched the corresponding RNA target, whereas only a background signal was observed in the negative control (“No Input” condition) or when the targeted allele was absent (Fig. 1b, c). We defined the delta of fluorescence as the amplitude between the lowest and the highest fluorescence values measured for each sample (Supplementary Fig. S1c).
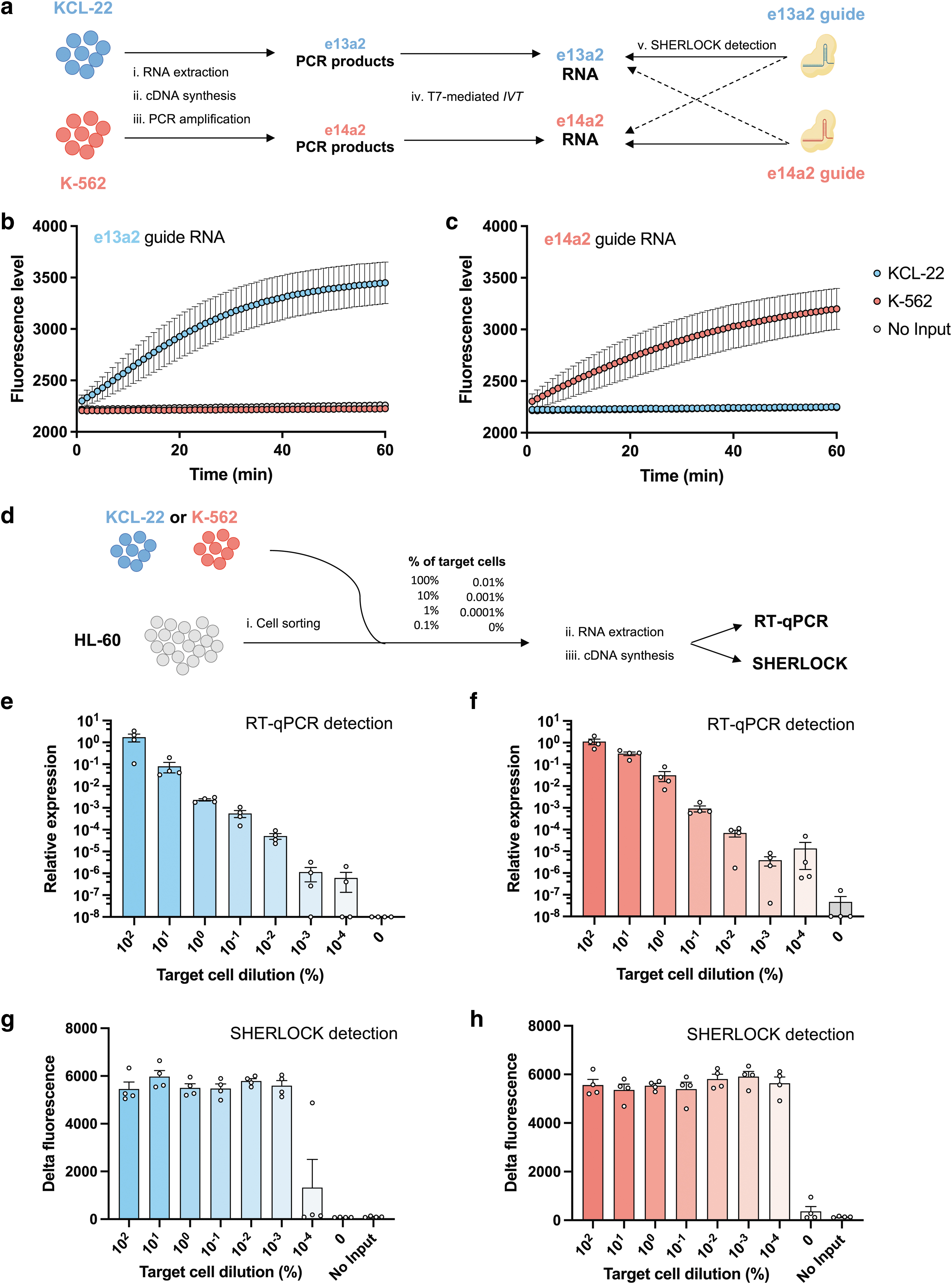
SHERLOCK enables the specific detection of BCR::ABL1 transcript variants.
With the e13a2 guide, the delta of fluorescence was 47-fold higher with e13a2 RNA products compared with e14a2 RNA products (Supplementary Fig. S3a). Similar profiles of amplitude and kinetic were obtained with the e14a2 guide with a 30-fold higher delta of fluorescence with e14a2 RNA as compared with the e13a2 products (Supplementary Fig. S3b). Overall, rapid and specific detection was observed for e13a2 guide and e14a2 guide in the presence of the target BCR::ABL1 variant compared with the nontargeted variant, suggesting specific recognition of BCR::ABL1 variants.
We next compared the sensitivity of RT-qPCR and SHERLOCK for the detection of BCR::ABL1 transcript. We performed BCR::ABL1 detection by cell spiking of known numbers of BCR::ABL1 positive cells (KCL-22 or K-562) into negative BCR::ABL1 cells (HL-60) (Fig. 1d). Samples were pretreated as they would be in clinic laboratories, the same cDNA samples being used as templates for both methods. As expected with the RT-qPCR, target cell concentrations were proportional to the levels of signal detection (Pearson R2 = 0.99 and 0.98) (Fig. 1e, f). By contrast, SHERLOCK detection plateaued without any correlation between the initial mutant cell number and the output signal (R 2 = 0.14 and −0.03) (Fig. 1g, h). RT-qPCR failed to detect one out of the four samples with the 10−3% dilution and two samples at the 10−4% dilution (Fig. 1e).
On the contrary, all samples were detected with SHERLOCK (Fig. 1g, h), except for the e13a2 lowest concentration (10−4%) where only one out of the four replicates was positive. At this dilution level, the heterogeneity of results could be explained by the absence or presence of the target in the input. We quantified BCR::ABL1 cDNA copies on cell spiking by RT-qPCR (see Materials and Methods section). The lowest concentration of e13a2 was not efficiently detected compared with the no target cell condition (0%) (Supplementary Table S4). This suggests that the lack of detection was due to the absence of target PCR products rather than reaching the sensitivity threshold of SHERLOCK detection.
We also confirmed targeted PCR amplification of BCR::ABL1 cDNA before SHERLOCK detection was needed to achieve high sensitivity. First, we showed that direct SHERLOCK detection on total RNA was unsuccessful (Supplementary Fig. S3e–g). When performing limited to moderate PCR amplification (using 5–25 cycles), we observed a relationship between the sensitivity and the number of PCR cycles (Supplementary Fig. S3g). In particular, detection was abrogated when limited amplification was performed before detection, confirming that efficient amplification is required for highly sensitive detection with SHERLOCK. Collectively, these results suggest that SHERLOCK enables robust detection of low concentrations of BCR::ABL1 with higher level of sensitivity than gold-standard RT-qPCR.
SHERLOCK enables the specific and sensitive detection of EGFRvIII intragenic rearrangement
To test the ability of SHERLOCK to detect oncogenic DNA rearrangements, we set up the detection of EGFRvIII intragenic rearrangement from GBM tumor samples (Supplementary Fig. S2c, d). For that, genomic DNA was extracted from a EGFRwt/vIII FFPE GBM tissue sample, and mutated DNA was amplified using allele-specific primers before SHERLOCK detection (Fig. 2a). EGFRwt alleles were amplified from DNA extracted from buffy coats of healthy donors. Using a EGFR vIII guide, highly specific detection was achieved in presence of EGFRvIII substrate (Fig. 2b).
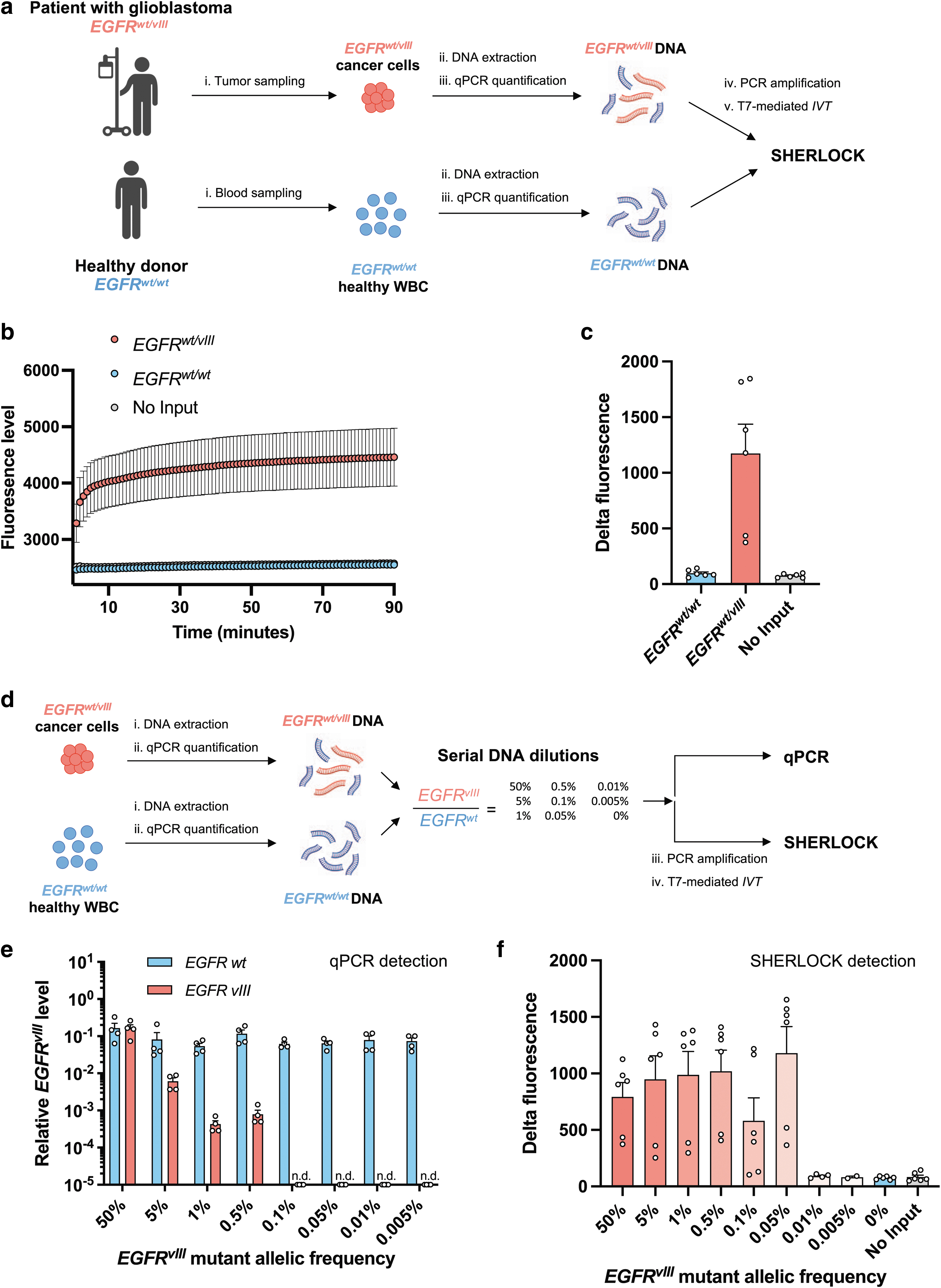
SHERLOCK enables the specific and sensitive detection of EGFRvIII rearrangement from patient samples.
Similar to the BCR::ABL1 detection, the delta of fluorescence was 12.1-fold higher with EGFRvIII compared with EGFRwt only (Fig. 2c). Next, we compared the sensitivity of detection of EGFRvIII by qPCR with SHERLOCK on the same samples of serial mutant DNA dilutions in wild-type (WT) DNA to mimic different mutant allele frequencies (Fig. 2e). With qPCR, efficient EGFRvIII detection was achieved from 50% to 0.5% of allele frequency (Fig. 2e). By contrast, SHERLOCK stopped detecting the EGFRvIII variant below 0.05% (Fig. 2f). Collectively, these results suggest that SHERLOCK is more sensitive than qPCR to detect EGFRvIII alleles.
SHERLOCK enables specific and sensitive detection of EGFRdel19 mutations
Next, we assessed whether SHERLOCK could be used for the detection of smaller oncogenic deletions that produce unique target sequences in the genome (Supplementary Fig. S2c, e). For this, genomic DNA was extracted from NSCLC tumor samples with the EGFRwt/del19 genotype, amplified using a nonspecific primer design (able to amplify both EGFRdel19 and EGFRWT alleles, Supplementary Table S1) and was subjected to SHERLOCK detection (Supplementary Fig. S4a, b, d). Using an EGFRdel19 guide, we achieved highly specific detection of the EGFRdel19 allele (Supplementary Fig. S4e).
Moreover, no significant difference was found between the EGFRWT DNA template and the no input condition delta of fluorescence (Supplementary Fig. S4f), highlighting the complete specificity of the assay. We next evaluated the sensitivity of SHERLOCK using serial DNA dilutions of EGFRdel19 and compared it with the ddPCR, routinely used for EGFR mutation detection in ctDNA from liquid biopsies of NSCLC patients. EGFRdel19 allelic frequencies were significantly detected down to 1% by SHERLOCK (Supplementary Fig. S4f), whereas ddPCR sensitivity reached 0.1% (Fig. 3e). These data suggest that nonspecific amplification of EGFR before SHERLOCK detection resulted in specific detection but limited sensitivity compared with ddPCR.

SHERLOCK enables the specific and sensitive detection of EGFRdel19 rearrangement from patient tumor samples.
Allele-specific SHERLOCK increased sensitivity of EGFRdel19 mutation detection
To enrich final PCR products with the EGFRdel19 alleles, we next used an allele-specific amplification of EGFRdel19 before SHERLOCK detection (allele-specific [AS]-SHERLOCK) (Supplementary Fig. S4a, c, d). An EGFRdel19 specific forward primer was designed to amplify EGFRdel19 only (Supplementary Table S1), whereas the PCR reverse primer and the guide RNA for detection remained unchanged. Using AS-SHERLOCK, the delta of fluorescence for the EGFRwt/wt was significantly higher than the negative control (Fig. 3b, c), possibly due to nonspecific amplification of the EGFRwt DNA (leading to the production of mutation containing amplicons).
However, the detection of EGFRwt/del19 was markedly more rapid and the delta of fluorescence was significantly higher than that of the EGFRwt/wt (Fig. 3b, c). We thus evaluated the sensitivity of AS-SHERLOCK approach using serial DNA dilutions of EGFRdel19 and compared it with the ddPCR. When using the allele-specific amplification, EGFRdel19 mutation was significantly detected with dilutions down to 0.05% (Fig. 3f). These results suggest that AS-SHERLOCK can provide a similar range of sensitivity to ddPCR for the EGFRdel19 mutation detection.
We next applied AS-SHERLOCK approach to the detection of EGFRdel19 in liquid biopsies from four NSCLC patients harboring an EGFRdel19 c.2236_2250 at diagnosis (Fig. 4a). Mutant ctDNAs were detected in three patients with ddPCR (Fig. 4b). Similar results were obtained using SHERLOCK confirming similar sensitivity for EGFRdel19 detection (Fig. 4c, d). In particular, high levels of fluorescence were obtained for the patient 3 providing robust confirmation of ddPCR results. For patient 2, which was negative for EGFRdel19 detection by ddPCR (Fig. 4b) and by NGS (not shown), we observed a higher delta of fluorescence than that of the EGFRwt/wt template (Fig. 4c, d). However, in absence of a positivity threshold determination, we cannot conclude on the patient's positivity on this sample (see Discussion section). Collectively, these results suggest that allele-specific SHERLOCK enables the specific detection of EGFRdel19 in patient samples with similar sensitivity than ddPCR.

AS-SHERLOCK enables specific and sensitive detection of EGFRdel19 rearrangement from patient plasma samples.
Discussion
Advances in targeted therapies have placed detection of nucleic acid biomarkers at the center of an increasing number of clinical and diagnostic applications. 2 Numerous cancers can be detected and monitored using PCR-based detection methods based on specific biomarkers that arise from gene rearrangements. In this study, we compared SHERLOCK performance with current PCR-based detection methods for BCR::ABL1 and EGFR mutations and provided evidence the first could be a valuable alternative. For the first time, large intragenic (i.e., EGFRvIII) and intergenic rearrangement (i.e., BCR::ABL1) were detected with SHERLOCK, adding a class of nucleic acid biomarkers to those previously detected with this tool.16,18–23
We first demonstrated that SHERLOCK enabled the specific detection of the two major isoforms of BCR::ABL1, EGFRvIII and EGFRdel. 19 We also compared the sensitivity of SHERLOCK for the detection of BCR::ABL1 and EGFR mutations to current PCR-based methods. For BCR::ABL1 mRNA detection, SHERLOCK demonstrated similar level of sensitivity to the gold-standard RT-qPCR. Interestingly, in the current settings we used, the SHERLOCK detection of the e13a2 isoform was slightly less sensitive than that of the e14a2. However, quantification of BCR::ABL1 cDNA copies confirmed that this difference in sensitivity is likely due to the absence of target products in the input (Supplementary Table S4).
In addition, even if equivalent number of target cells has been used to compared e13a2 and e14a2 detection sensitivity, the absolute number of BCR::ABL1 mRNA per cell might differ between the KCL-22 and K-562. Indeed, e14a2 isoform expression was reported to be higher than e13a2 counterpart in patient samples providing potential explanation of sensitivity difference observed in this study (Fig. 1g vs. Fig. 1h). 26 Sensitivity differences in the two BCR::ABL1 isoforms could also be explained by differences in the amplification efficiencies and in the RNA guide cleavage performance.
Collectively, it suggests that SHERLOCK enables highly sensitive detection of large oncogenic rearrangement and, therefore, constitutes a valuable tool for minimal residual disease monitoring. For CML patients, frequent relapse are observed after TKI cessation in patients who had negative BCR::ABL1 RT-qPCR for at least 2 years. We speculate negative SHERLOCK detection of BCR::ABL1 can be envisioned as an additional parameter to meet treatment stop criteria.
At the same time, we observed improved sensitivity for the detection of EGFRvIII and EGFRdel19 DNA mutations with SHERLOCK or AS-SHERLOCK, compared with qPCR and ddPCR, respectively.
We demonstrated that SHERLOCK sensitivity is tightly linked to the specificity of the initial PCR amplification step. As illustrated by the EGFRdel19 SHERLOCK assay (Supplementary Fig. S4), co-generation of PCR products that are not substrates for SHERLOCK detection reduces the assay sensitivity. This is particularly a challenge for the detection of rare smaller rearrangements in which the WT and mutated alleles can be amplified by the same PCR primers. Under these conditions, PCR will preferentially amplify the wild-type allele, thus reducing the likelihood of the mutant-specific crRNA subsequent detection.
This explains the higher sensitivities obtained by SHERLOCK assays for viral or bacterial sequence detection, amplification of these sequences being completely specific of the pathogen. To bypass this limit, we combined an allele-specific amplification step with the SHERLOCK detection, to enrich PCR products in mutated sequences and facilitate CRISPR-Cas13 activation. Despite a reduced specificity, probably linked to PCR amplification from WT sequences, this approach was more sensitive than the conventional SHERLOCK assay (Fig. 3f and Supplementary Fig. S4f).
As AS-SHERLOCK generated some background signal with WT control, the interpretation of the sample positivity needs a positivity threshold determination, not performed in our study (i.e., patient 2; Fig. 4c). An extensive method validation is required before any implementation in clinic (according to the ISO15189 standard). Even if optimizations are still needed, particularly regarding assay specificity, AS-SHERLOCK could provide a new sensitive method to monitor minimal residual disease in NSCLC patients through the detection of the EGFRdel19 mutation in ctDNA.
The current PCR-based detection methods of biomarkers (RT-qPCR, qPCR, and ddPCR) rely on specialized instrument, trained personnel, and expensive reagents. However, one of the major challenges of the coming decades is to ensure equitable access to medicine. The need for better medicine access in emerging economic regions (1.8 million people are diagnosed each year with lung cancer worldwide), 27 and the increasing prevalence in developed countries (from 70,000 CML patients in 2010 in the United States to 180,000 expected in 2050) 28 will stress health care systems.
This requires the use of inexpensive, mostly equipment-free techniques that can be performed ideally without trained personnel. 12 CRISPR-based diagnostics methods have recently received increasing attention during the SARS-Cov2 outbreaks, with the development of versatile point-of-care tests (POCTs).18,20,21 CRISPR-based POCTs offer rapid, low-cost, instrument-free, and deployable detection of nucleic-acid biomarkers by combining isothermal amplification, CRISPR-based detection of target of interest, and user-friendly readout.
Conclusions
The evolution of treatment toward personalized medicine is revolutionizing cancer management. The precise identification of specific tumor-related nucleic acids will be needed to monitor treatment effectiveness and residual disease. In this study, we demonstrate that CRISPR-Cas13a enables rapid and sensitive detection of oncogenic rearrangements both in cell lines and patient samples. SHERLOCK has the potential to outperform the gold-standard PCR-based methods for the detection of BCR::ABL1 fusion oncogenes and EGFR oncogenic mutations. Future optimizations could even lead to one-step and instrument-free detection assays, allowing the development of handy POCTs.
Footnotes
Acknowledgments
We thank Omar Abudayyeh and Jonathan Gootenberg (McGovern Institute for Brain Research, Massachusetts Institute of Technology, Cambridge, MA) for sharing precious advices and help on SHERLOCK technology and Emmanuel Tetaud (MFP, UMR CNRS 5234, University of Bordeaux, France) and Fanny Boissier (Darfeuille Lab, UMR CNRS 5320/Inserm U1212, University of Bordeaux, France) for their helpful discussion and assistance on biochemical aspects of Cas13a activity. We thank Claudine Chollet (Laboratory of Hematology, CHU Bordeaux, France) for the quantification of BCR::ABL1 copy number. The authors thank the staff of FACSility and CRISP'edit, technology platforms (INSERM US 005—CNRS UAR 3427—TBMCore, University of Bordeaux, France) for assistance.
Authors' Contributions
Conceptualization (equal), investigation (lead), visualization (lead), writing—original draft (lead), and writing—review and editing (equal) by G.C. Conceptualization (equal), funding acquisition (lead), investigation (lead), resources (lead), supervision (supporting), visualization (supporting), writing—original draft (equal), and writing—review and editing (equal) by S.A. Investigation (equal) and writing—review and editing (equal) by L.K. and J.R. Conceptualization (equal), investigation (equal), resources (lead), writing—original draft (equal), and writing—review and editing (equal) by V.P.-M. Writing—review and editing (equal) by L.B., S.Du., A.B., and F.M.-G. Conceptualization (supporting), resources (supporting), and writing—review and editing (equal) by D.C. Conceptualization (equal), funding acquisition (lead), supervision (lead), writing—original draft (equal), and writing—review and editing (equal) by S.Da. and B.T.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by Ligue contre le Cancer—Comité des Landes, France Intergroupe de la Leucémie Myéloïde Chronique (Fi-LMC), Association Française Pour la Recherche sur le Cancer du Pancréas (AFRCP) and Novartis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.