
Abstract
The CRISPR-associated Cas12b system is the third most efficient CRISPR tool for targeted genome editing in plants after Cas9 and Cas12a. Although the genome editing ability of AaCas12b has been previously investigated in rice, its off-target effects in plants are largely not known. In this study, we first engineered single-guide RNA (sgRNA) complexes with various RNA scaffolds to enhance editing frequency. We targeted EPIDERMAL PATTERNING FACTOR LIKE 9 (OsEPFL9) and GRAIN SIZE 3 (OsGS3) genes with GTTG and ATTC protospacer adjacent motifs, respectively. The use of two Alicyclobacillus acidoterrestris scaffolds (Aac and Aa1.2) significantly increased the frequency of targeted mutagenesis. Next, we performed whole-genome sequencing (WGS) of stably transformed T0 rice plants to assess off-target mutations. WGS analysis revealed background mutations in both coding and noncoding regions with no evidence of sgRNA-dependent off-target activity in edited genomes. We also showed Mendelian segregation of insertion and deletion (indel) mutations in T1 generation. In conclusion, both Aac and Aa1.2 scaffolds provided precise and heritable genome editing in rice.
Introduction
Cas12b (formerly known as C2c1) is a type V-B CRISPR-Cas endonuclease system that can efficiently introduce dual-RNA-guided DNA double-strand breaks (DSBs) in both mammalian1,2 and plant genomes.
3
Cas12b effector proteins originated from thermophilic bacteria Alicyclobacillus spp. (AaCas12b and AacCas12b) and Bacillus spp. (BthCas12b and BhCas12b) showed editing activities in high temperature range (48–50°C) in vitro, while Alicyclobacillus acidiphilus
While the 5′-TTN-3′ protospacer adjacent motif (PAM) was sufficient for optimal cleavage activity of AaCas12b in mammalian cells, -VTTV- with a preference for 5′-ATTV-3′ and 5′-GTTG-3′ PAMs was strictly related to higher genome editing frequencies in rice. 3 Based on the comparison of Cas12b orthologs, AaCas12b has been found to be superior to Aac- and Bth- in genome editing in rice with up to 20% mutation frequency in protoplast cells. 3 The versatility of Cas12b in plant cells has also been explored by developing transcriptional activation and repression vector systems created based on two orthologs (Alicyclobacillus acidoterrestris -Aac and A. acidiphilus -Aa) and several sgRNA scaffold combinations. 3 Cas12b is one of the most precise endonucleases among the other genome-editing tools due to its low tolerance to single-base mismatch within the seed region,1,4 which presumably limits its off-target activities throughout the genome.
In plants, while the efficacy of CRISPR technologies has been largely demonstrated and to a considerable extent remains under constant improvement, their safety in crops needs to be evaluated systematically. The 20-nt protospacer and a PAM adjacent to the target sequence in the genome tightly control the targeting specificity of Cas-endonucleases, yet off-target cleavage activity may still occur within the genome other than the target sequence. The position of mismatches with protospacer, particularly the proximity to PAM has been found critical, while off-target could still occur with ∼6-mismatches between the protospacer and genome. 5
Plant transformation often involves the tissue culture process known to create background mutations, which are not necessarily harmful, and in most cases, variations arising from these events will have no effect on the phenotype. For example, several hundred such somaclonal variation mutations originated from tissue culture per plant were observed during Cas12a genome editing in rice, but only a fraction of them were associated with coding sequences. 6 Off-target mutations can occur across the entire genome by a given sgRNA, while DSBs can cause cellular toxicity through activation of DNA damage regardless of the genomic region. Currently, whole-genome sequencing (WGS) is the only method to distinguish genome-wide off-target activity from somaclonal variation and spontaneous mutations.6,7
Off-target effects of Cas9- and Cas12a-mediated genome editing have been previously demonstrated in Arabidopsis thaliana, 8 tomato, 9 cotton, 10 grape, 7 and rice.6,11 Cas9 and Cas12a have been found to be very specific nucleases to induce desired changes in crop plants. 6 CRISPR-Cas12b systems have been demonstrated for genome editing in multiple plant species, including rice, 3 Arabidopsis, 12 and cotton. 13 However, there is no comprehensive genome-wide analysis for investigating off-target mutations caused by Cas12b in plant genomes.
Here we designed a study to assess putative off-target mutations generated by Cas12b genome editing in stably transformed rice plants. To maximize the efficiency of multiplex genome-editing, we first evaluated mutation frequencies of five different vectors constructed based on a previously established CRISPR/AaCas12b system including dual-polymerase II (Pol II) promoter expression system and hammerhead virus–hepatitis delta virus (HH-HDV) dual-ribozyme-based sgRNA processing.3,14 These vectors contained one protospacer for each of the OsEPFL9 and OsGS3 target sites, engineered with RNA scaffolds and transcriptional activation domains (TV, 15 TV-MS2-VPR 15 , and Act3.016). We have obtained up to 75% on-target indel mutation frequency in multiplex-edited plants by an Aac scaffold. Six independent T0 edited plants, along with control plants, were selected for WGS to discover mutations arising from genome editing reagents and the tissue culture process.
Materials and Methods
Plasmids and vector construction
T-DNA vectors were constructed based on Gateway cloning as previously described. 3 We first generated pYPQ141-ZmUbi-RZ entry clones carrying AaCas12b-sgRNA 1.2, sgRNA 3.8, AacCas12b-sgRNA, Aac.3, and Aac.4 scaffold sequences separately (Supplementary File S1). The protospacer targeting OsEPFL9 and OsGS3 were synthesized as single oligonucleotides (designated as L10 and L12, respectively), which were phosphorylated, annealed, and ligated into pYPQ141-ZmUbi-RZ (Addgene No. 86196) at the BsmB I sites. 17 For constructing multiplex vectors carrying sgRNA 1.2, sgRNA 3.8, and AacCas12b-sgRNA scaffolds, HH-scaffold-L12-HDV- cassette was introduced into the PstI-BamHI sites on previously formed entry vectors.
For Aac.3 and Aac.4 scaffolds, cassettes were cloned using HIFI primers (Supplementary File S1). The sgRNAs in entry vectors were confirmed by Sanger sequencing. AaCas12b expression vector pYPQ292 (Addgene No. 129672), multiplexed sgRNA entry clones, and pYPQ203 (Addgene No. 86207) destination vector were used for the three-way Gateway assembly to generate the T-DNA vectors. Final vectors were confirmed by restriction digestion. Maps of entry plasmids and T-DNA vectors can be found in Supplementary File S2.
Protoplast assay
Rice (Oryza sativa L.) Japonica cultivar Kitaake was grown on ½ MS basal salt medium in the dark at 28°C for 14 days. Etiolated rice shoots were used for protoplast isolation. Thirty to 40 healthy rice seedlings were cut into ∼0.5–1.0 mm strips using razor blades. The strips were then transferred into a 90 mm Petri dish with 8–10 ml of enzyme solution (1.5% cellulase R10, 0.75% macerozyme R10, 0.6 M mannitol, 10 mM MES at pH 5.7, 10 mM CaCl2, and 0.1% BSA), followed by vacuum-infiltration for 30 min in the dark using a vacuum pump at −15 to −20 in.-Hg, and then incubated at 60–80 rpm for 7–9 h at 28°C in the dark. The digested products were filtered through a 40 μl cell strainer on a Petri dish and transferred into a sterile 50 ml Falcon tube.
The protoplast pellets were collected by centrifugation at 100 g for 5 min and suspended with 10 ml of W5 buffer (0.5 M mannitol, 20 mM KCl and 4 mM MES at pH 5.7) for washing twice. Protoplasts were then examined and counted under a microscope. Then the protoplasts were collected again by centrifugation at 100 g for 2 min and then suspended in MMG buffer (0.4 M mannitol, 15 mM MgCl2, and 4 mM MES at pH 5.7) at a concentration of 2 × 106 cells ml−1. For transformation, a volume of 200 μl of protoplasts was gently mixed with 30 μl of plasmid DNA (1000 ng/μl; prepared by Qiagen Midiprep Kit) and 230 μl of PEG transformation buffer (40% w/v PEG4000, 0.2 M mannitol, and 0.1 M CaCl2).
Thirty microliters of plasmid expressing green fluorescent protein (GFP) (2x35S:GFP) was used as a control to calculate the transfection efficiency. After a 30-min incubation at room temperature, the transformation reaction was stopped by adding 900 μl of W5 buffer. Protoplasts were collected by centrifugation at 250 g for 5 min and resuspended in 1 ml of W5 solution. The resuspended protoplasts were transferred to 12-well plates and incubated in the dark for 48 h at 32°C. After 16 h of incubation, transformation efficiency was calculated based on the expression of GFP under fluorescence microscopy.
Stable transformation
Agrobacterium-mediated transformation was performed as described previously. 6 The binary vectors used in this study were transformed into Agrobacterium tumefaciens strain EHA105 using electroporation. For rice transformation, dehusked seeds were sterilized with 70% ethanol for 1 min and washed one time with sterile water. These seeds were further sterilized with 50% sodium hypochlorite for 30 min on a shaker. Seeds were then washed with sterile distilled water five times. Finally, the sterilized seeds were dried on a sterilized filter paper and cultured on solid callus induction medium and incubated at 28°C in a growth chamber for 2–3 weeks. Actively growing calli were collected for subculture at 28°C for 1–2 weeks. Agrobacterium cultures were collected and resuspended in liquid infection medium (OD600 = 0.4) containing 100 μM of acetosyringone.
Rice calli were immersed in the Agrobacterium suspension for 10 min. These calli were then dried on sterilized filter paper and plated on a coculture medium at 25°C in a dark growth chamber for 3–5 days. The infected calli were moved to a sterile plastic bottle and washed five times with infection medium with 100 mg/L of timentin to remove excess bacteria. After being dried on a sterilized filter paper, these calli were transferred onto a selection medium containing 50 mg/L of hygromycin and 100 mg/L of timentin at 30°C in a growth chamber for 4 weeks. After the selection stage, actively growing and regenerating calli were moved onto a regeneration medium (RegI) at 30°C with a 16-h light/8-h dark cycle. Transgenic seedlings were then transferred to RegII medium and grown for 2–3 weeks before being transferred into the soil.
Next-generation sequencing analysis for editing efficiency in protoplasts
To measure the editing efficiencies, PCR amplicons from protoplasts were subjected to next-generation sequencing (NGS). Pairs of primers to amplify each target region harboring the protospacer sequences (L10 and L12) were designed by the addition of three different barcodes to the 5′ ends (Supplementary File S1). The resulting PCR amplicons of pooled gDNA were analyzed by agarose gel electrophoresis and column purified by the QIAquick PCR purification kit. Samples were arranged as a total of ≥1.5 μg (20 ng/μl) in concentration and subjected to Illumina HiSeq2500 for sequencing (Azenta, Inc., South Plainfield, NJ). The resulting data were analyzed by CRISPRMatch 18 and CRISPResso2. 19
Genotyping of genome-edited plants
Genomic DNA was isolated from the leaves of 30 stable transgenic T0 rice plants grown on regeneration media according to the CTAB method. 20 To assess genome editing at two sites, PCR amplicons produced by first-round PCR with the primers specific to OsGS3 and OsEPFL9 genomic sequences were amplified with barcoded Hi-TOM 21 primers in the second-round PCR (Supplementary File S1). PCR amplicons were then concentrated as 20 ng/μl for sequencing using Illumina HiSeq2500 (Azenta, Inc.). Sequencing data were then sorted by CRISPRMatch 18 and analyzed by HiTom software 21 and CRISPResso219 programs. For genotyping of T1 generation, Hi-TOM-based NGS was applied as described above. A chi-square test was performed to analyze the heritability of mutations in assessed T0 lines.
WGS and data analysis
For WGS, three transgenic lines with high efficiency of indel mutations for both target sites were selected for each sgRNA scaffold. The genotypes of edited and control plants for WGS are described in Figure 4A. Genomic DNA (gDNA) was extracted from 50 mg leaves by Qiagen (DNeasy Plant Pro Kit) as described by the manufacturer. 1.6 μg of gDNA was used to construct sequencing libraries for WGS, which was provided by Azenta, Inc. All individual samples were sequenced at a depth of average 35 × with the average coverage being 98%. The WGS analysis was done similar to our previous articles.6,22
In brief, adapters and low-quality reads were filtered using SKEWER (v. 0.2.2), 23 and trimmed reads were mapped to the japonica variety Nipponbare genome (MSU7) with BWA (v. 0.7.17) mem algorithm. 24 Samtools (v. 1.9) 25 was used to generate sorted and BAM files. Picard was applied to mark duplicated reads and the Genome Analysis Toolkit (GATK v. 3.8) 26 was used to realign the reads near indels. After the above processing, relevant BAM files were generated and used to detect whole-genome mutations. Single-nucleotide variants (SNVs) were detected by LoFreq (v. 2.1.2), 27 MuTect2, 28 and VarScan2(v. 2.4.3), 29 and INDELs detected by MuTect2, 28 VarScan2 (v. 2.4.3), 29 and Pindel (v. 0.2). 30 To obtain high-confidence mutations, only mutations identified in all three software were retained for subsequent analysis. Potential off-target sites were predicted by Cas-OFFinder (v. 2.4) 31 by allowing up to 5-nt mismatches.
Data analysis and figure plotting are completed with Python and R. The WGS data have been deposited in the Sequence Read Archive in the National Center for Biotechnology Information (NCBI) under the accession number BioProject PRJNA810274.
Results
Comparison of sgRNA scaffolds for AaCas12b-mediated multiplexed genome editing in rice protoplasts
A previous study demonstrated that fusing transcription activators to Cas9 can boost its editing activity by modulating chromatin accessibility. 32 Whether this works for Cas12b or not remains unclear. In this study, we designed five CRISPR-Cas12b systems (pLR4112 to pLR4116) containing AaCas12b or AaCas12b-activator (AaCas12b-TV, AaCas12b-TV-MS2-VPR, and AaCas12b-Act3.0) coupled with five different guide RNA scaffolds, including Aac, 3 Aa1.2, 3 Aa3.8,1,3 Aac.3, 16 and Aac.416 (Fig. 1A–C and Supplementary File S1). No transcription activator was fused or recruited for both vectors pLR4112 and pLR4113, representing controls. For vectors pLR4114 to pLR4116, activation domains/systems such as TV, 15 VPR, 3 and Act3.016 were directly fused to AaCas12b or recruited by guide-RNA scaffolds (Fig. 1A–C). To simultaneously express two sgRNAs for each vector, we used the HH-HDV double-ribozyme system for precise processing of sgRNA excision (Fig. 1B).

Comparison of sgRNA scaffolds for AaCas12b genome editing in rice cells.
The editing efficiencies of engineered vectors with various sgRNA scaffolds were assessed at two target sites (OsEPFL9 and OsGS3) in rice protoplasts. Cas12b and corresponding sgRNAs were delivered as assembled T-DNA plasmids to rice protoplasts by PEG-mediated transformation. Transformed cells by a GFP vector as negative control did not show any editing activity. We observed genome editing at various levels by different scaffold-carrying constructs at both target sites (Fig. 1D, E). At the OsEPFL9 site, 7.2% and 7.9% editing efficiencies were obtained with Aac and Aa1.2 scaffolds, respectively, which were significantly higher than Aa3.8 and Aac.4 scaffold vectors (Fig. 1D). Similarly, higher editing efficiencies at the OsGS3 site have been obtained by Aac and Aa1.2 scaffold vectors as 13.8% and 18.87% (Fig. 1E). In general, Aa3.8, Aac.3, and Aac.4 scaffold vectors displayed relative low editing efficiencies for both OsEPFL9 (∼5%) and OsGS3 (0.65% to 2.1%) sites.
Among total mutations, indel mutations on target sites were predominant (Fig. 1D, E). Most of the indel mutations that occurred were deletions and the frequency of deletions was slightly higher in OsGS3 than OsEPFL9 (Supplementary Fig. S1). This difference was more pronounced in the case of the Aa1.2 scaffold vector. AaCas12b mainly generated 8–13 base pair (bp) deletions at the target sites (Fig. 2A). The deletions occurred about 14–23 nucleotides distal to the PAM sites (GTTG for OsEPFL9 and ATTC for OsGS3) by Aac scaffold (Fig. 2B) and 12/14–24 nucleotides distal to PAM sites for Aa1.2 scaffold (Fig. 2C). Overall, our data suggest that Aac and Aa1.2 scaffolds mediated better editing efficiencies in targeting both genomic loci. Therefore, we proceeded to use pLR4112 (Aac-carrying) and pLR4113 (Aa1.2-carrying) vectors for evaluating the on- and off-target effects of AaCas12b in stably transformed rice plants.
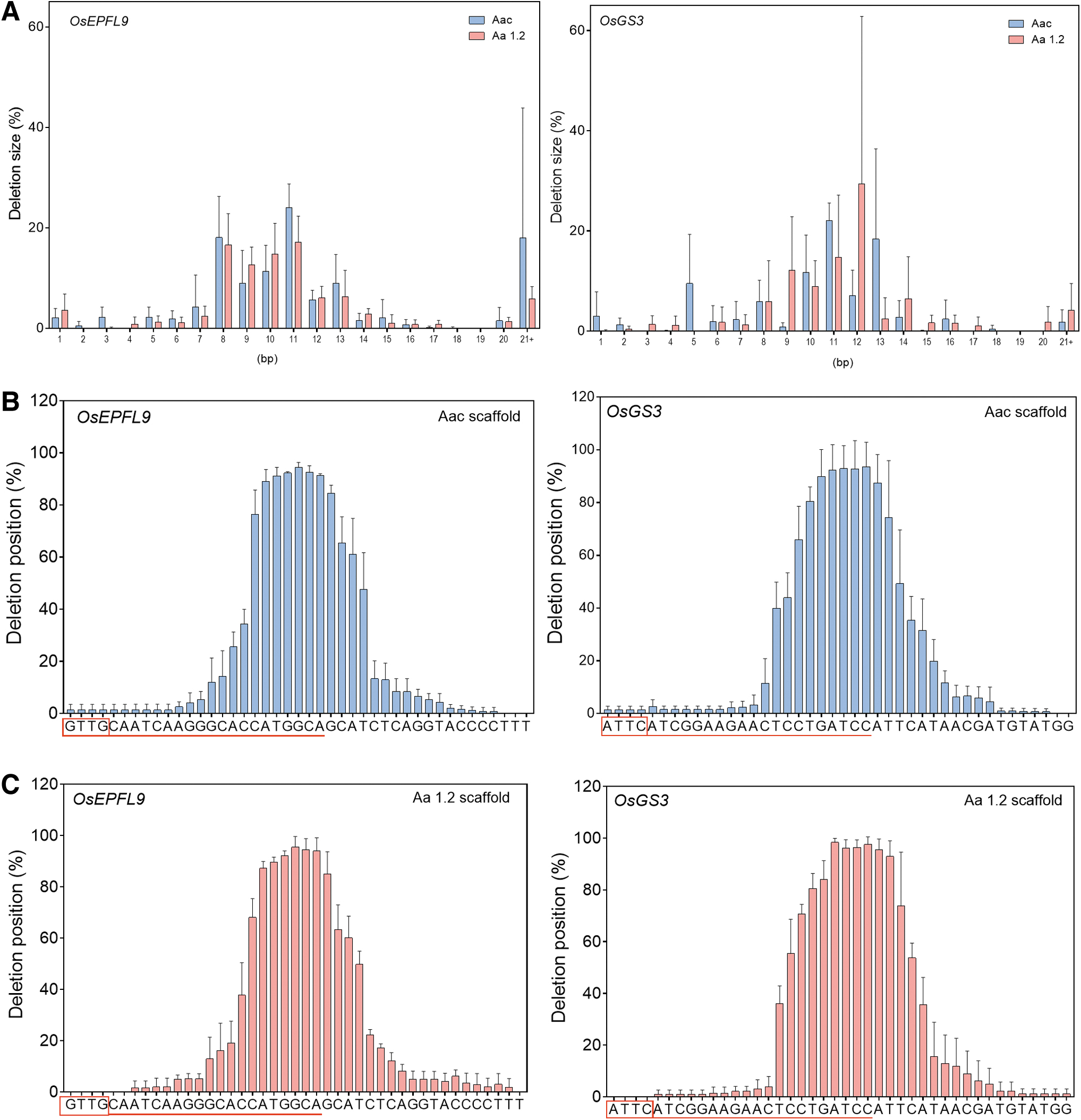
Deletion profiles by different AaCas12b systems in rice cells.
Cas12b genome editing in stably transformed rice plants
Based on the initial comparison of scaffold vectors, Aac and Aa1.2 were used to generate stably transformed rice plants by Agrobacterium-mediated transformation. We generated 64 and 156 transgenic plants from Aac and Aa1.2 scaffold vectors, respectively. Genotypic evaluation of 30 T0 plants and the distribution of indel frequencies by NGS of PCR amplicons showed that the OsGS3 site was edited in higher ratios compared with OsEPFL9 (Fig. 3A). With the Aac scaffold vector, we obtained 18 mutant plants for OsEPFL9 and 26 mutant plants for OsGS3 sites, both including 2 biallelic plants. In the case of the use of Aa1.2 scaffold, OsEPFL9 site was edited at a lower frequency with the same vector (10 mutants in 30 plants). The total mutation rate with the same scaffold was up to 96% in OsGS3-edited plants (Fig. 3B).
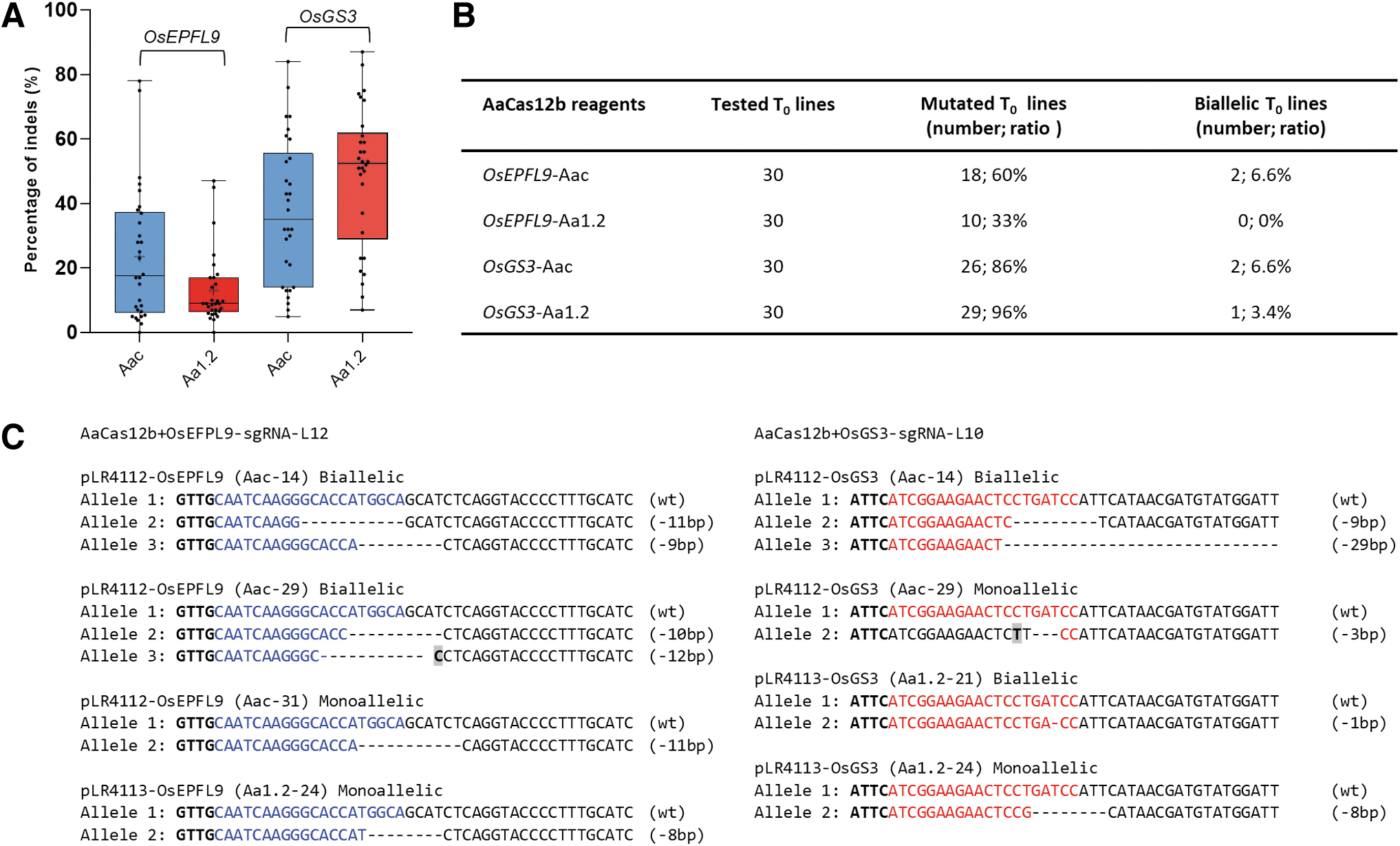
Cas12b-mediated multiplexed editing in stable T0 lines.
A total of five plants carried biallelic mutations with varying sizes of deletions (Fig. 3C). To assess the off-target effects of AaCas12b-expressing and genome-edited plants, we selected plants with high indel frequencies for both target sites. Indel frequencies varied between 0–78% for OsEPFL9 and 4.9–84% for OsGS3 using the Aac scaffold in edited plant lines (Supplementary Fig. S2). For the Aa1.2 scaffold vector, indel frequencies were between 0–47% for OsEPFL9 and 11–87% for OsGS3 sites (Supplementary Fig. S2). Nine plants were further evaluated by Sanger sequencing on both target sites.
When these lines were examined for their zygosity states, one plant (Aac-14) was shown to carry biallelic mutations for both target genes, while Aac-29 and Aa1.2–21 were biallelic for only one gene (Fig. 3C). The rest of the lines carried heterozygous, biallelic, or chimeric combinations for two target sites based on the evaluation of mutant reads by NGS data (Fig. 3C). 33
Validation of on-target mutations by WGS
To evaluate the on-target and potential off-target effects of Cas12b systems in rice, we exploited five T-DNA constructs including two dual-sgRNA constructs to target two genes (Fig. 1C). According to the genotyping results, two plants from Aac- (14 and 29) and three plants from Aa1.2 scaffolds (4, 21, and 24) were selected for WGS analysis. By our experimental design that included genome-edited lines and no-sgRNA controls, we were able to analyze background mutations caused by tissue culture and Agrobacterium-mediated transformation (Fig. 4A). To ensure high confidence on base calling, all 13 individual plants were sequenced at 32–58 × (average ∼48 × ) in-depth (Supplementary Table S1) and a stringent mutation mapping and calling pipeline was developed for WGS analysis (Fig. 4B). Figure 4C shows the on-target analysis of six plants by WGS at two genomic sites.
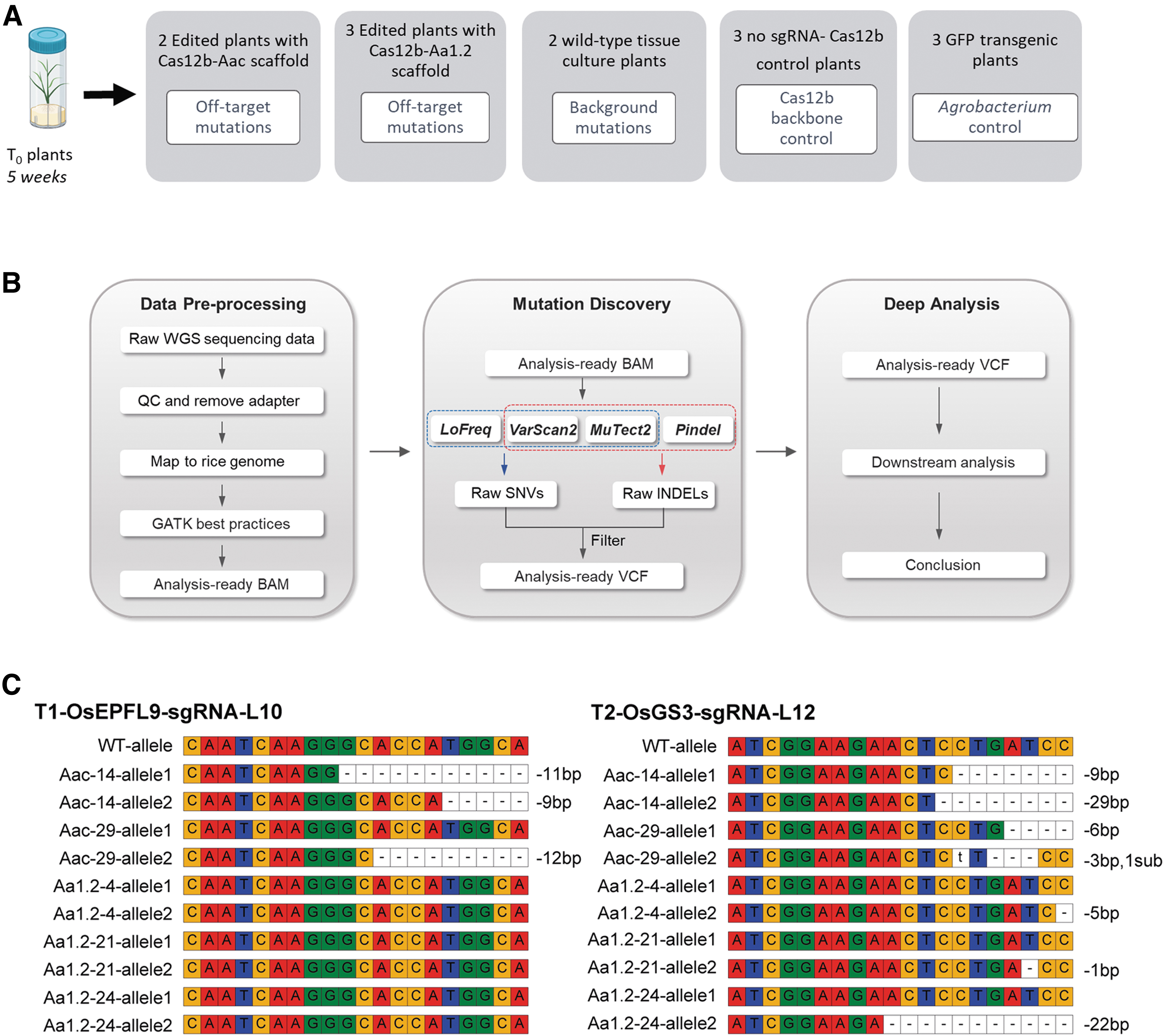
WGS analysis for on- and off-target analyses of Cas12b-mediated genome editing in rice.
For the OsEPFL9 site, the two Aac scaffold lines carried the biallelic allele, while none of the Aa1.2 scaffold lines showed mutations at this site (Fig. 4C). At the OsGS3 site, all T0 plant lines carried deletions with deletion sizes varying between 1 and 29 bp. In general, the data were consistent between amplicon sequencing and WGS. Based on WGS, Aac-14 and Aac-29 plants carried alleles with 9, 11, and 12 bp deletions at the OsEPL9 site, which were consistent with the previous amplicon sequencing analysis. However, we also discovered some discrepancies between these two genotyping methods. Plant lines denoted as Aa1.2-4 and -21 carried edited OsEPFL9 alleles at a very low frequency based on the previous amplicon sequencing. No indels were detected at this target site in both lines by WGS. Aa1.2-24 had -8 bp indel that showed 42% mutation frequency by amplicon sequencing, but this mutation could not be validated by WGS.
On the contrary, we detected a 7 bp deletion in the Aac-29 line by WGS, which was not detected by amplicon sequencing previously. We reasoned these differences might be due to different sensitivity levels of two sequencing technologies and/or different parts of the same plants being sampled in these two independent experiments.
Detection of off-target mutations by WGS
The assessment of sgRNA-independent mutations is important for a genome editing tool such as CRISPR-Cas12b. WGS data generated from 13 plants of different groups allowed us to uncover the genome-wide distribution of small mutations. We performed variant calling using a standardized bioinformatic pipeline for all samples (Fig. 4B). Three variant-calling software programs were used to identify SNVs and small insertions and deletions (indels), with high-confident variants shared by all software. Mutations with frequencies below 10% may not be called out, as such low-frequency mutations may have arisen from sequencing errors. T0 lines carried varying numbers of SNVs and indels according to WGS analysis when those were mapped to the reference rice genome. The numbers of SNVs in Cas12b-edited lines on average (∼374 for Aac and 410 for Aa1.2) were slightly higher than wild-type (∼176) and GFP-transformed plants (∼233) (Fig. 5A).
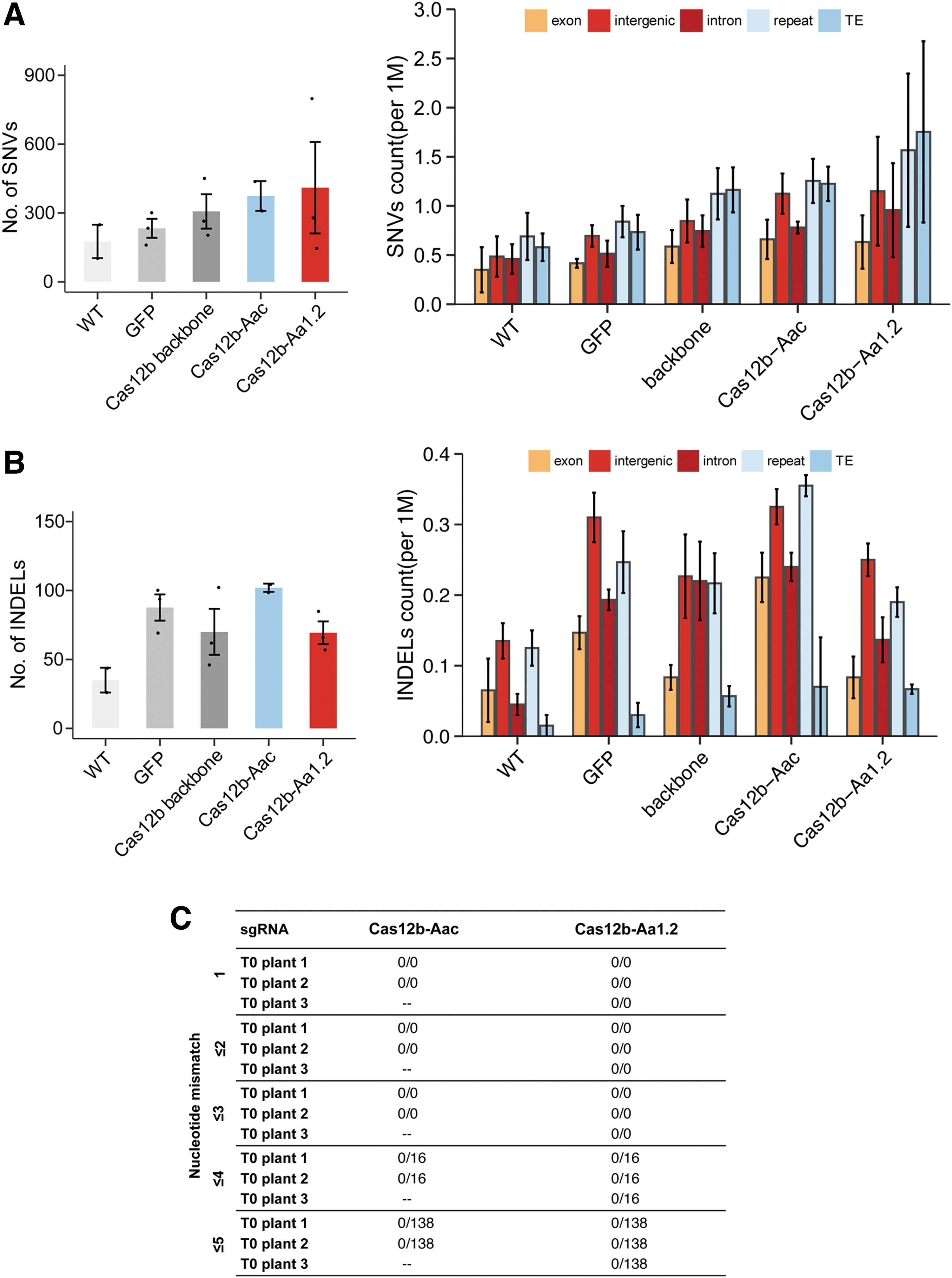
Off-target analysis by WGS.
Further analysis showed that SNVs have been found mostly in transposable elements and repeat regions in the genome, while their occurrence was rare in coding sequences (Fig. 5A, right graphic). The number of indels was 100 per genome on average among edited lines and control groups (Fig. 5B). Moreover, the number of SNVs and indels observed is consistent with the range of other research groups and our previous studies,6,34–37 suggesting that these mutations were derived from tissue culture. Intergenic regions and transposable elements harbored more indels than other genomic regions in wild-type, GFP transformants, and edited plant lines. The overall genome-wide distribution of SNV and indel mutations in both Aac and Aa1.2 scaffold-edited lines had similar patterns to the GFP and wild-type control plants (Supplementary Fig. S3).
In addition, we detected a total of 18 T-DNA insertion events in T0 lines (Supplementary Fig. S4). Insertions dispersed over nine chromosomes except Nos. 8, 9, and 12. Two GFP insertion events denoted as GFP_1 and GFP_2 were found with three and two copies, respectively. Other insertions identified by WGS include two copies of Cas12b-Aac_2 and four copies of backbone sequences. All these analyses strongly suggest the SNVs and small indel mutations in these genome-edited T0 lines are mostly background mutations caused during tissue culture and Agrobacterium-mediated transformation.
To identify protospacer-dependent off-target mutations in T0 plants, we analyzed the specificity of sgRNAs with the Cas-OFFinder software. The program identified potential off-target sites by searching the whole rice genome for similar sequences to protospacers allowing 1 to 5 mismatches in sequence alignments (Fig. 5C). With the criterion allowing 1-nt to 3-nt mismatches in the protospacer, both protospacers for the two target sites with Cas12b-Aac and Cas12b-Aa1.2 had predicted no off-target sites, suggesting the high uniqueness of these two target sites in the rice genome. However, with 4-nt and 5-nt mismatches, Cas-OFFinder predicted 16 and 138 off-target sites in the rice genome, respectively.
However, we have previously shown that 2-nt mismatch is enough to largely kill the genome editing activity by AaCas12b in rice. 3 Indeed, when we mapped these potential off-target sites to the selected Aac- and Aa1.2 lines with WGS data, no mutation was observed at any of these putative off-target sites.
Transmission of targeted mutations to the next generation
T1 plants from two lines (Aac-14 and Aac-29) were generated to assess both transmissions of edited mutations from T0 and the segregation of the CRISPR/AaCas12b transgene. We used NGS of PCR amplicons to genotype these lines. Almost all plants carried mutated alleles defined in T0 in varying ratios in the T1 generation. We have observed only one additional editing event of a large deletion (-91 bp) at the OsGS3 site of the progeny of Aac14 (data not shown). Since these plants did not carry AaCas12b cassette in Aac14-T1 lines, this additional mutation might have occurred in T0 plants and transmitted to the T1 generation.
We observed enlarged seed size in three heterozygous lines (Aac-18, Aa1.2-24, and Aa1.2-30) for OsGS3 among 13 T0 lines evaluated as 0.75–0.82 cm in width compared with wild type (∼0.7 cm). Based on zygosity types of mutations described previously, 33 segregation ratios in T1 were in accordance with the Mendelian ratio (1:2:1) in all four cases except the Aac-29 plants at the OsEPFL9 site (p ≤ 0.05) (Table 1). Hence, targeted mutations in rice by CRISPR/AaCas12b are readily transmissible to the next generation.
p values are calculated using the two-sided Chi-square test, ns, not significant.
CH, chimeric; HT, heterozygous; HM, homozygous; WT, wild type.
Discussion
CRISPR-Cas endonucleases in both natural and engineered variant forms are fundamental tools for genome engineering in plants.38,39 As a newly emerging system, Cas12b orthologs have been comprehensively investigated for genome editing considering different PAM requirements for optimal mutagenesis and targeted gene activation and repression. 3 Structural studies demonstrated that genome editing efficiency depends on the interaction between guide-RNA and Cas9. 40 Modifications in functional modules proximal to 3′ end of tracrRNA demonstrated that distinct parts of the secondary structure of sgRNA directly affect DNA cleavage and determine orthogonality in different systems. 41 Scaffolds can be engineered through the A-U flip, stem extension, and RNA aptamer modifications. 42
In this study, we successfully expressed different sgRNA-scaffolds in rice cells and compared the targeting efficiency of AaCas12b with modified scaffolds. An Aac scaffold carrying vector (pLR4112) was used as a control in our study as it previously demonstrated comparable editing frequencies with Aa1.2 and Aa3.8 at four targeted sites with different PAMs. 3 In addition to these systems, we combined transcription activators/systems, TV, VPR, and Act3.0 with AaCas12b cassette, to explore their possible effects on chromatin opening and improving targeting efficiency. Liu et al 32 reverted a potent dCas9-TV activator 15 to a nuclease-active Cas9-TV and showed it significantly improved Cas9-mediated genome editing in rice genome regardless of genomic chromatin state, suggesting a chromatin modulation activity. CRISPR/AaCas12b-Act3.0 system used the MS2 RNA aptamer in the sgRNA scaffolds to enhance gene activation by recruiting more copies of activation domain 2 × TAD. 16 Harnessing nuclease active AaCas12b and transcription activators might improve genome editing in rice, as was shown earlier. 32
However, based on our protoplast assay, none of the tested transcription activators TV, TV-MS2-VPR, and the Act3.0 system coupled with the AaCas12b endonuclease improved targeting efficiencies in rice. Rather, reduced genome editing efficiency was observed at two independent target sites (Fig. 1D, E). A major factor for this result may be that the new conformations of these hybrid proteins (Cas12b-TV or Cas12b-TV-MS2-VPR) affect the catalytic nuclease activity. Cas12b also evolved differently than Cas9 and Cas12a through its unique mechanism of RNA/DNA recognition and cleavage. 43 Noncoding RNA aptamers were previously used to recruit effector proteins to enhance activation/repression studies in mammalian cells. 44 While these configurations may not necessarily affect CRISPR DNA targeting, it may interfere with DNA cleavage that requires additional CRISPR/Cas conformational changes upon DNA binding. Supporting this notion, our study here showed that the combination of sgRNAs with different scaffolds and the recruitment of transcription activators could negatively interfere with the overall genome editing efficiency of AaCas12b. Nevertheless, we compared Aac and Aa1.2 scaffold carrying constructs and found that the Aac scaffold facilitated more robust Cas12b-mediated multiplexed genome editing than the Aa1.2 scaffold in rice stable lines. Hence, we recommend the use of Aac scaffold to facilitate AaCas12b-mediated genome editing in plants.
While on-target mutations can be identified by amplicon sequencing, WGS is required for the analysis of off-target effects of CRISPR/Cas reagents in plant genomes.6,7,22 Our WGS analysis demonstrated that both wild-type and transformed plant lines carried varying numbers of SNVs and indels (up to 249 SNVs and 44 indels in wild-type and 801 SNVs and 105 indels in edited lines) distributed throughout the rice genome. While spontaneous mutations caused by seed propagation contribute only a small amount of this variation, these discovered mutations are largely somaclonal variations. 6 The random distribution of these mutations such as in transposable elements and repeat regions is consistent with our previous findings.6,34
Efficient targeting with CRISPR reagents in plant genomes with minimal off-target effects is not only important to establish a convenient system for genome editing, but also ensures the safety of resulting food products. Having a different cleavage mechanism than Cas9 and some similarities to Cas12a, AaCas12b possesses a potential for both non-homologous end joining-mediated targeted mutagenesis and homology-directed repair-mediated targeted gene replacement and insertion studies in plants. Previously, off-target effects of a version of Bacillus hisashi Cas12b were assessed in mammalian cells using a biased method, Guide-Seq. 2 BhCas12b was found to be very specific compared with SpCas9 when tested at multiple target sites. To our knowledge, the use of WGS for the assessment of off-target activity by a Cas12b nuclease has not been previously done in a plant genome. With WGS, we did not detect protospacer-dependent or protospacer-independent off-target effects of AaCas12b in the rice genome.
Our results demonstrate that AaCas12a-expressing T0 plants, despite carrying somaclonal variation due to tissue culture, are devoid of off-target mutations. It suggests that transgenic AaCas12b plants could be safely used for facilitating other genome editing delivery approaches such as virus delivery. 45 Finally, our T1 generation analysis provided novel information on the nature of AaCas12b activity in progeny and the stability of the transmission of mutations in plants.
Conclusion
In this study, we created and tested a series of constructs with modified sgRNA scaffolds/systems to improve AaCas12b genome editing efficiency in plants. We have obtained relatively higher mutation efficiencies with Aac and Aa sgRNA 1.2 at two loci that encode agronomically important traits, while other modifications reduced sgRNA targeting efficiency. This study showed that scaffold sequences and associated effector proteins may affect Cas12b enzymatic activity. We demonstrated that transgenic AaCas12b rice plants do not possess genome-wide off-target effects, and the on-target mutations are largely germ line transmittable. Our study paves the road for efficient and safe use of CRISPR/AaCas12b system for crop improvement.
Footnotes
Authorship Contribution Statement
Y.Q. and F.G. conceived the project and designed the experiments. F.G. constructed the plasmids and generated stable lines. F.G. and G.L. analyzed the NGS data. C.P. provided vectors with transcription activators. F.G. and Y.C. performed the protoplast assay. Y.W. and T.Z. analyzed the WGS data. F.G. and Y.Q. wrote the article with input from all other authors.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the USDA-NIFA Biotechnology Risk Assessment Research Grants Program (no. 2020-33522-32274) and the NSF Plant Genome Research Program (nos. IOS-1758745 and IOS-2132693).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.