
Abstract
The recently established prime editor (PE) system is regarded as next-generation gene-editing technology. This methodology can install any base-to-base change as well as insertions and deletions without the requirement for double-stranded break formation or donor DNA templates; thus, it offers more targeting flexibility and greater editing precision than conventional CRISPR-Cas systems or base editors. In this study, we introduce the basic principles of PE and then review its most recent progress in terms of editing versatility, specificity, and efficiency in mammals. Next, we summarize key considerations regarding the selection of PE variants, prime editing guide RNA (pegRNA) design rules, and the efficiency and accuracy evaluation of PE. Finally, we highlight and discuss how PE can assist in a wide range of biological studies and how it can be applied to make precise genomic corrections in animal models, which paves the way for curing human diseases.
Introduction
CRISPR-Cas9, derived from the adaptive immune system in prokaryotes, has been repurposed for genome engineering purposes in a wide range of living organisms. 1 The Cas9 nuclease recognizes a target sequence under the guidance of a single-guide RNA (sgRNA) and induces a double-stranded break (DSB) three bases upstream of a protospacer adjacent motif (PAM) sequence.2,3 The DSB triggers either a cellular nonhomologous end-joining (NHEJ) DNA repair pathway or a homology-directed repair (HDR) pathway. The former generates small insertions and deletions (indels) that can disrupt gene function, whereas the latter results in precise edits when a donor repair template exists.4,5 NHEJ is active all the time, but HDR occurs only during the G2 and S phases of the cell cycle, which limits its use to actively dividing cells only.6–9
To our knowledge, published examples of in vivo HDR in adult mammals to date have reported relatively low editing efficiencies of ∼0.1–6.5%. 10 In addition, the introduction of a DSB before HDR is often associated with undesirable side effects such as indel formation,6,11 DNA translocation, 12 large deletions, 13 and p53 activation.14,15 These shortcomings might, in part, be overcome by the use of base editing systems, which have recently been developed to effectively modify single nucleotides without breaking the DNA double strand. To date, three types of base editors, including cytosine base editors (CBE), 16 adenine base editors (ABE), 17 and glycosylase base editors (CGBE),18–20 have been established, which mediate C-to-T, A-to-G, and C-to-G base substitutions, respectively, in mammalian cells. In principle, these can reverse >70% of the pathogenic point mutations in humans. 21
The basic principle behind base editors is the fusion of a DNA-modifying enzyme with a catalytically disabled Cas protein, and so, they can be used in both dividing and nondividing cells. In addition, with the application of phage-assisted continuous evolution, structure-guided design, and machine learning, the editing efficiency of base editors is currently ∼50% for CBE, 22 ∼40.87–64.22% for ABE, 23 and ∼15% for CGBE in mammalian cells. 24 This is ∼10-to-100-fold higher efficiency than HDR in obtaining the desired mutations.25,26 However, although the most recently developed base editors act more efficiently and generate fewer indel by-products, they are more restricted in the generation of bystander off-target modifications, and the inability to install targeted nucleotide transversions (except C-to-G transversion), deletions, and insertions.21,27
Fortunately, these limitations of the base editor have recently been solved by the development of a prime editor (PE). This is a recently developed genome editing technology, which enables unprecedented precision in the installation of any moderately sized genetic variation without the need for a double-strand DNA break, and it thus offers greater versatility than CRISPR nucleases and base editors. 28 With the aid of PE, >89% of known human disease-causing genetic variants can in theory be corrected. 28
Prime editing combines a Cas9-H840A nickase (nCas9)–reverse transcriptase (RT) fusion protein with a prime editing guide RNA (pegRNA) containing the desired editing sequence (Fig. 1A). The pegRNA consists of three elements: (1) a protospacer sequence of 20 nucleotides (nt), which specifies the target site and guides the Cas9-H840A to cut the PAM-containing strand to form a single-strand DNA (ssDNA) flap; (2) a primer binding site (PBS), which serves as the template for the RT; and (3) the RT template (RTT), which contains the desired edition at the target site (Fig. 1A). 28 When the protospacer sequence in the pegRNA binds to DNA, the PAM-containing strand of the DNA is only cut by the Cas9n three bases upstream from the PAM to generate an ssDNA flap. The PBS then binds with the DNA flap and serves as a primer for RT, which incorporates nucleotides that contain the desired edit into the 3′ flap.
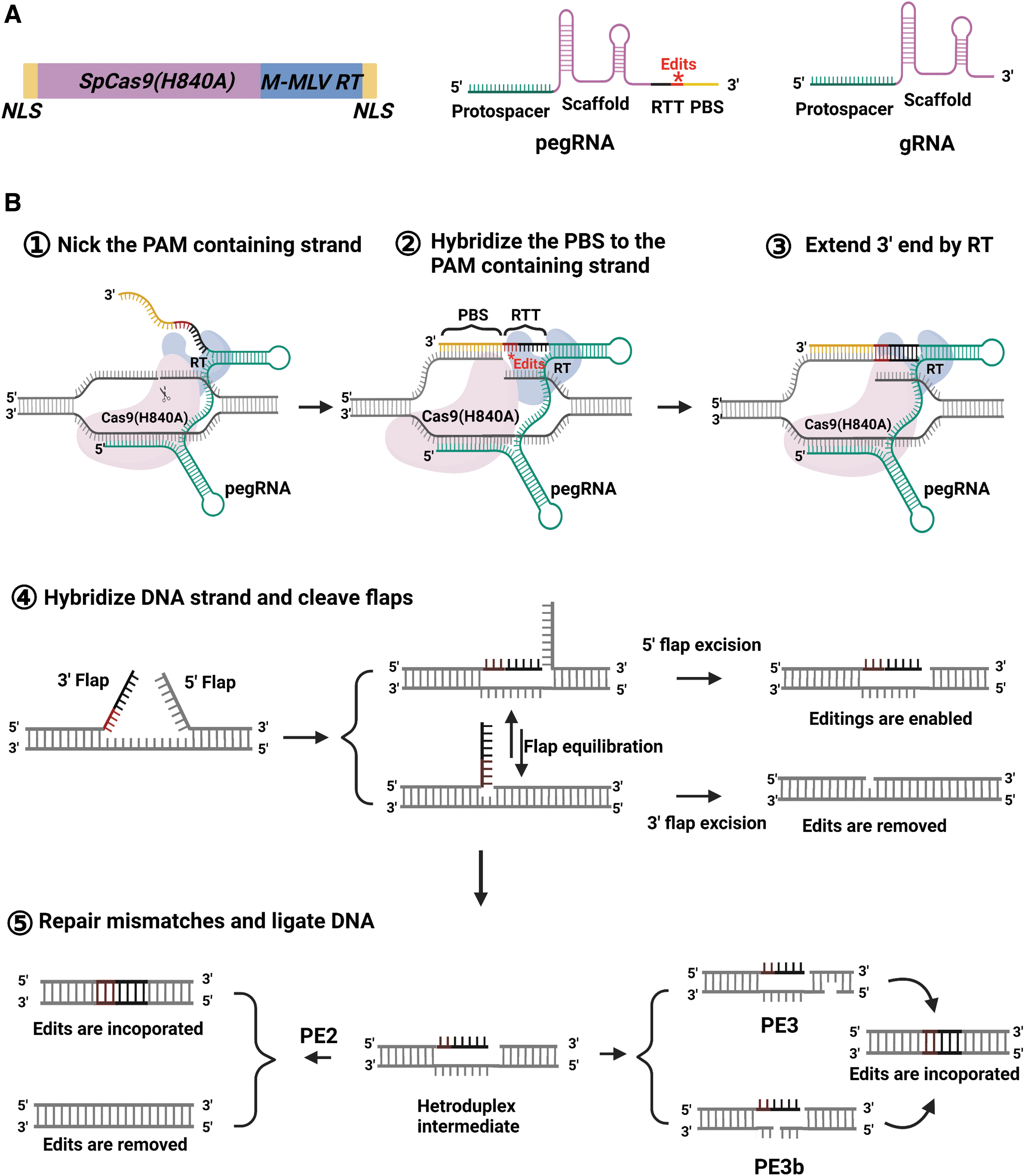
The basic principle of prime editing.
After the reverse transcription process, the nicked double-stranded DNA hypothetically undergoes an equilibration between the edited 3′ flap and the unedited 5′ flap. The cleavage of the unedited 5′ flap by a 5′endonuclease (e.g., FEN1) or by a 5′exonuclease (e.g., EXO1) then leads to the desired edit.29,30 The first generation of the prime editor (PE1) consisted of a wild-type Moloney murine leukemia virus (M-MLV) RT attached to the C-terminus of H840A nickase. To increase the polymerase activity of PE1, a second-generation prime editor (PE2) was produced by incorporating five mutations in the RT to enhance its DNA binding affinity. Compared with PE1, PE2 could achieve up to a 5.1-fold increase in editing efficiency when producing indels at target sites.
However, the editing efficiency of PE2 is compromised because the incorporated edit in the PAM-containing strand has a 50% possibility of being cut out during the DNA mismatch repair (MMR) and ligation process (as shown in Fig. 1B). Thus, to further optimize PE2, a PE3 system was designed, which utilizes a simple sgRNA to create a second nick in the nonedited strand. This stimulates the resynthesis of this nonedited strand by using the edited strand as the template through the endogenous MMR system. Although this secondary nicking can result in a fully edited duplex, it sometimes leads to indels if the edited strand is nicked before the RTT sequence has been incorporated. The PE3b system solves this problem by using a second guide (also known as nicking guide RNA [ngRNA]) against the anticipated edit, such that the nick on the nonedited strand only occurs after successful sequence incorporation on the edited strand (Fig. 1B).
Using this approach, PE3b results in 13-fold fewer indels when compared with PE3 in human cells. However, in other species, PE3 and PE3b are not always more efficient than PE2. 28 For example, they have a similar editing efficiency to PE2 in plants and zebrafish.31–33 It has been suggested that the differences in the RT reactions and DNA repair pathways among these different species might account for this discrepancy.28,33–35
Guide RNA (gRNA)-dependent and gRNA-independent off-target effects are major factors that influence the successful application of CRISPR-based genome editing tools.36,37 Such off-target effects can result from similarities between on-target and off-target sequences, and the overexpression of functional elements.38,39 Conventional CRISPR-Cas9 relies on NHEJ or HDR to repair the break and facilitate gene editing, whereas base editors enable single-nucleotide conversion without causing DSB. However, the latter might lead to gRNA-independent off-target editing in both DNA and RNA because of the intrinsic affinity of deaminases to RNA and single-stranded DNA. 38 Although the PE system does not rely on either NHEJ or HDR, it still requires other intrinsic repair pathways (such as MMR), to be sufficiently active in cells, but these pathways are responsible for the gRNA-dependent off-target effects observed.40,41
However, PE generates fewer off-target mutations than Cas9 nucleases. This might be because two additional DNA hybridization events (i.e., RT priming and DNA flap resolution) beyond Cas9 binding are required. Each of these provides an additional checkpoint alongside the spacer sequences to reject off-target sequences. 39 In addition, guide-independent off-target mutations or other types of genetic alterations such as telomere integrity, endogenous retroelements, and gene splicing are not observed by applying the M-MLV RT moiety.28,42
From the start, the versatility, accuracy, and moderate efficacy of prime editing have provided exciting prospects for biomedical studies and clinical applications. However, at the earliest stages of development, it did not exhibit the same wide editing versatility as CRISPR-mediated HDR, which can achieve up to 2 kb insertion and 10 kb deletion,43,44 or the robust editing efficiency of base editors. 10 Therefore, in this review, we first consider these two drawbacks of PE as major guidance cues to examine its recent progress in achieving a more extensive editing versatility, better specificity, and higher efficiency in mammals. Next, since the pegRNA sequence is more complex than canonical sgRNA, we summarize several crucial pegRNA design rules and web-based design tools that make PE more feasible for use in scientific research.
We also describe methods that support on-target and off-target evaluation of PE and emphasize how this technique can be used to mediate precise genomic corrections in animal models and thus promote biological studies. Finally, we discuss the future directions and perspectives of PE, and how this technology might pave the way for curing human diseases.
Improving the Editing Versatility of PE
PE has been demonstrated to be capable of installing insertions of up to 44 base pairs (bp) and deletions of up to 80 bp in human cells, with high ratios of desired edits in the by-products. 28 However, for the replacement or deletion of large sequences (i.e., >100 bp), the efficiency of the original PE was very low. However, by the application of pairs of pegRNAs, five independent research groups, respectively, achieved the deletion, replacement, integration, and inversion of large sequences, or chromosomal translocation, although with slightly different strategies (Fig. 2A, B and Table 1).45–50
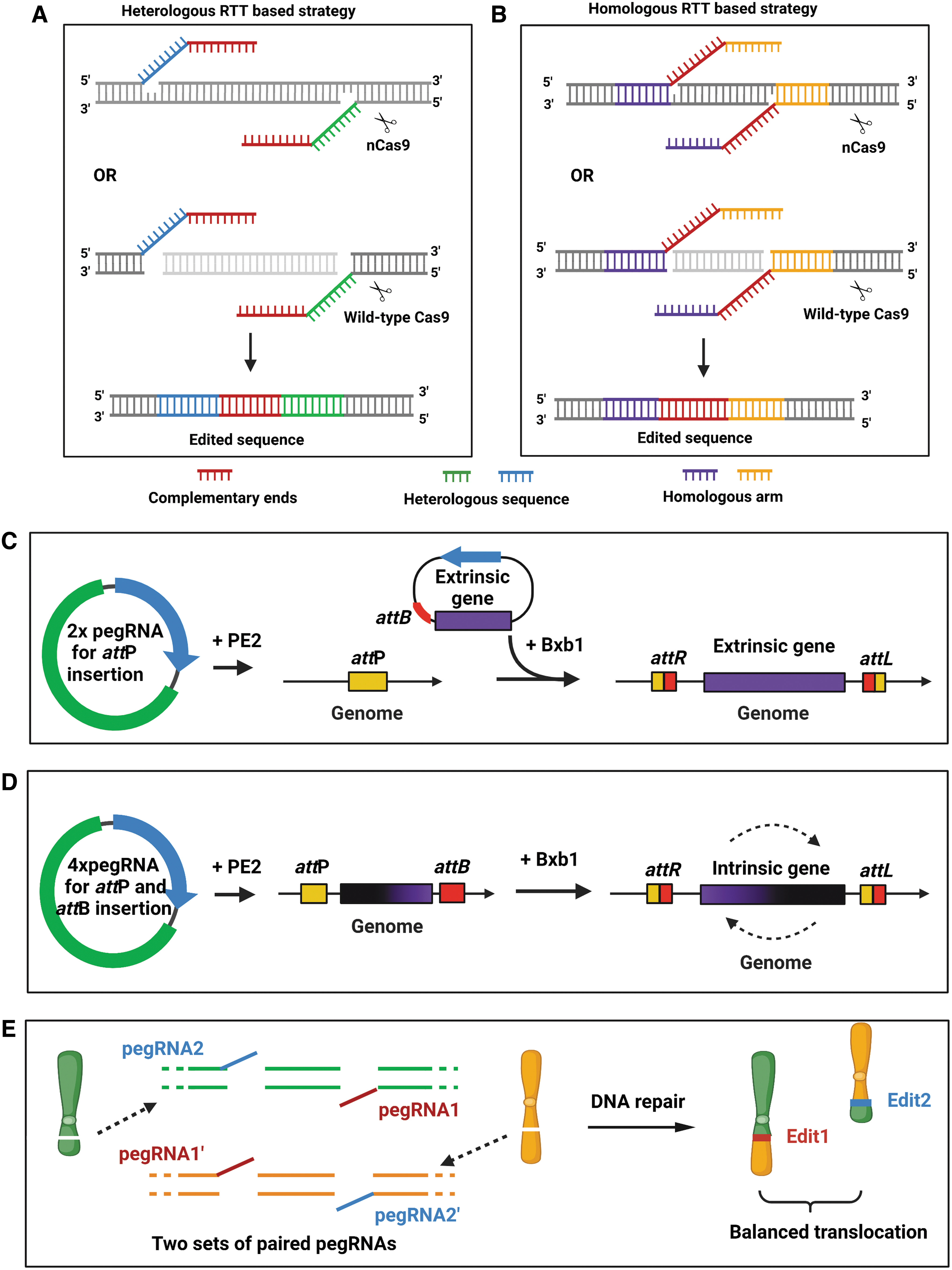
Expanded prime editing versatility through paired pegRNA. Schematic illustration of
Large-sequence modification strategies based on prime editor
Bi-PE, bidirection prime editing; bp, base pairs; GRAND, genome editing by RTTs partially aligned to each other but nonhomologous to target sequences within duo pegRNA; indels, insertions and deletions; NA, not available; PEDAR, PE-Cas9-based deletion and repair; RTT, reverse transcriptase template; twinPE, twin prime editing; WT-PE, wild-type Cas9 based prime editor.
For example, Wang et al, Jiang et al, and Anzalone et al each designed a pair of pegRNAs with no homology (or heterology) to the original DNA (Fig. 2A), and they named their strategies as follows: GRAND editing (genome editing by RTTs partially aligned to each other but nonhomologous to target sequences within duo pegRNA), PEDAR (PE-Cas9-based deletion and repair) editing, and twinPE (twin prime editing), respectively,45–47 although their working principles were almost the same. After DNA nicking or cleavage by nuclease, each pegRNA contains editing template sequences that reverse transcribe two ssDNAs that are partially complementary to each other and thereby resolve the 3′ flap. This 3′ flap annealing maintains its stability by preventing nuclease degradation.
In addition, the newly synthesized DNA strands are highly dissimilar to the endogenous target site and therefore avoid hybridizing to the original DNA sequence. These two unique properties of nonhomologous pair pegRNAs guarantee their high efficiency. After equilibration between the edited and original strands, the original strands are cleaved, and the edited strands are repaired through gap-filling. Ligation of the pair of nicks results in the replacement of the endogenous sequence with the paired 3′ flap sequences. GRAND editing and twinPE utilize Cas9 nickase (nCas9) and can achieve up to 1.3 kb DNA sequence deletion and up to 1 kb DNA sequence insertion at the same time.
In contrast, PEDAR editing utilizes a fully active Cas9, and can achieve up to 10 kb DNA deletion, although the simultaneous DNA insertion length is short (i.e., just ∼60 bp). This discrepancy is likely due to wild-type Cas9 being more efficient at introducing larger deletions and having faster DSB repairing kinetics, than it does with the RT process. 45 Furthermore, with twinPE, the preinsertion of Bxb1 attP sequences at safe harbor loci of the genome might facilitate the knockin of an attB-containing DNA donor plasmid with a sequence >5 kb in the presence of serine recombinase (Bxb1) (Fig. 2C). In line with this, when the target sequence is flanked by the twinPE-mediated multiplex insertion of attP and attB, the activity of Bxb1 results in an inversion of the target sequence up to 40 kb (Fig. 2D).
As GRAND, twinPE, and PEDAR use pegRNA RTT with DNA heterologous to the genome, these editing methods offer more template sequence flexibility and have greater potential to make larger insertions with a relatively shorter RTT. However, all of these strategies are accompanied by DNA fragment insertion when they are being applied for sequence deletion. To achieve precise deletion without insertion, Choi et al developed PRIME-Del editing, through which two templated 3′ flap sequences (which are initiated by a pair of pegRNAs) are, respectively, homologous to the downstream and upstream regions of two nick sites generated by pegRNA. After DNA repair, these 3′ flap sequences are incorporated into the target site and accomplish precise deletion (Fig. 2B). 48 However, when the short sequence is placed 5′ to the deletion-specifying homology sequences, PRIME-Del can perform simultaneous deletion and short insertion (Fig. 2B).
As described above, homologous and heterologous RTT-based strategies for large-sequence manipulations have their own characteristic advantages. However, for large-sequence deletions or chromosomal rearrangements, Tao et al conjugated wild-type Cas9 to the RT and established a so-called wild-type Cas9 based prime editor (WT-PE) system. 49 They showed that a pair of pegRNAs with a homologous region in-between the extended 3′ DNA flap and the PAM at the proximal DSB end outperformed a pair of pegRNAs with a heterologous region to the same target genome in terms of editing efficiency, although both methods could achieve megabase-scale sequence deletion and chromosomal translocation (Fig. 2E). 49 Later, this group further established the bidirection prime editing (Bi-PE) system, which used nCas9 at this time, and was also based on the principle of paired pegRNAs as described above; similarly, they demonstrated that the homologous RTT-based strategy outperformed the heterologous one in both efficiency and accuracy when large-sequence manipulations were executed. 50
Although these two pegRNA strategies can mediate long-sequence modifications, and therefore offer more advantages than HDR in terms of efficiency and specificity, long-length pegRNA pairs need to be cloned, and this is more challenging technically than cloning conventional sgRNA pairs. In addition, the presence of appropriate PAM sequences oriented toward each other at opposing strands of the target genomic sequence is the prerequisite for genomic modification by the pegRNA pair. However, this strict sequence context requirement might limit their optimal application. Fortunately, the development and optimization of several Cas variants with different PAM specificities would relax this constraint.51–53 To increase the PAM availability when adopting SpCas9-based PEs, various SpCas9 variants were designed to contain H840A mutations before conjugation to M-MLV RT, to develop PE2 variants. These have been shown to relax the canonical PAM preferences but with fewer off-target effects than the corresponding SpCas9 variants. 54
The SpCas9 variants include the SpCas9-VQR and SpCas9-VRQR variants for NGA PAM 55 ; the SpCas9-VRER variant for NGCG PAM 56 ; the SpCas9-NG and SpG variants for NG PAM51,57; and the SpRY variant for a near-PAM-less sequence. 51 In parallel, an orthogonal SaCas9 nickase or SaCas9-KKH nickase is fused with the M-MLV RT in PE2 to develop orthogonal PEs, Sa-PE2, and Sa-KKH-PE2, which recognize an NNGRRT and NNNRRT PAM, respectively, and provide an appreciable genome editing efficacy and accuracy, broadening the toolkit of available PE systems. 58
Enhancing the Specificity and Efficiency of PE
PE offers great flexibility for introducing a variety of different types of sequence alterations into the genome, however, one drawback of the original PE was its limited application and unsatisfactory efficiency when compared with the base editors. 59 Since its initial development in 2019, much effort has been made to boost the efficacy of PE and maintain or even reduce its off-target effects. Because PE makes use of the CRISPR nuclease, M-MLV RT, and pegRNA, and its activity relies on the MMR machinery in the host, it is expected that any factors that can optimize these various functional elements will improve the efficacy of the overall editing technique. In the following sections, we describe these contributory factors in further detail.
Optimization of the PE protein derived from the original PE architecture
The nuclear localization of PE is essential for its gene-editing activity. However, some PE2 is known to leak out of the nucleus, which might compromise its performance. 58 The incorporation of an additional c-Myc NLS at the N terminus of PE, and a variant bipartite SV40 NLS (vBP-SV40) and SV40 NLS at the C terminus, leads to a new PE variant, named PE2*, which has been shown to achieve nearly complete nuclear localization in U2OS and HeLa cells. 58 In addition, the fusion of some amino acid peptides, such as an N-terminal peptide from NFATC2IP (NFATC2IPp1) and a phosphomimetic peptide from IGF1 (IGF1pm1), to the N-terminal PE2 would increase the PE2 protein level probably through increasing PE2 translation. 60
To open the chromatin structure around the target site, two chromatin-modulating peptides (CMPs), called the high-mobility group nucleosome binding domain 1 (HN1) and histone H1 central globular domain (H1G), respectively, can be added to the N-terminus and C-terminus of nCas9 to generate CMP-PE-V1. 61 When this is combined with a proximally binding dead sgRNA (dsgRNA), it can achieve up to 3.92-fold higher editing efficiency than PE3. 61 Recently, Liu and colleagues updated PE2 to PEmax by optimizing the M-MLV RT codon; introducing SpCas9 R221K and N394K double mutations; adding another C-terminal c-Myc NLS; and including a 34-aa linker containing a bipartite SV40 NLS between the nCas9 and M-MLV RT. 41 They showed that PEmax outperforms PE2* and CMP–PE–V1 in HeLa cells. However, it is interesting to note that inserting CMPs into PEmax decreases rather than increases their activity, and so, it is recommended that for now it is better to replace PE2 with PEmax in most PE applications. 41
The specificity of PE can be improved by replacing SpCas9 H840A with its high-fidelity variants, including EvoCas9, 62 HypaCas9, 63 Cas9-HF, 55 eSpCas9, 64 and SniperCas9. 65 Although all of these PE2 variants exhibit high specificities compared with the classic PE2, only eSp-PE2 and Sniper-PE2 maintain a comparable editing efficiency, whereas PE2-HF, Hypa-PE2, and Evo-PE2 exhibit dramatically low on-target activity. 66
Improvement of the expression efficacy of the PE system
PE has a higher efficiency in HEK293T cells (which are easily transfected) than in other cell types such as HeLa, K562, and U2OS cells. This suggests that the robust expression of PE in host cells might influence their activity, and that nontransfected cells might impair the overall PE performance.28,29,67,68 To enhance the expression of PE in cells, species or tissue-specific promoters that drive Cas9 and M-MLV RT moiety can be used2,69–71; for example, hepatocyte-specific promoters, including TBG, TTR, and PAH, are used to obtain liver-restricted transgene expression.72–74 RNA polymerase III (pol III) promoters such as U6 are commonly used for the expression of small-noncoding RNAs.
Robust pegRNA expression driven by the U6 promoter could be achieved by the following methods. (1) Adding an extra G in the first position of the pegRNA and ngRNA sequences might boost their expression if the first nucleotide is not a G. This is because a G residue in the first position of the gRNA promotes U6 promoter-driven RNA transcription.2,75,76 (2) Doubling the number of pegRNA expression cassettes and using two promoter systems to drive pegRNA expression can enhance pegRNA expression. 77 (3) Mutating the fourth uracil among successive uracils into cytosine in the pegRNA scaffold erases a putative transcription termination signal, and thus increases the expression of pegRNA. 78
However, the versatility and specificity of pegRNA might be sacrificed by the above approaches. For example, adding an extra G to the 5′ end of pegRNA or gRNA might impair the RNA-DNA heteroduplex and affect Cas9 fidelity; the pol III promoters hardly tolerate more than four contiguous U residues, which makes PE fail to correct poly-thymine [poly-(T)] containing targets. Luckily, these restrictions are recently relieved by pegRNA initiated through the RNA polymerase II (pol II) promoters. 79 In addition to the PE system itself, the toxicity and efficiency of the transfection agents utilized might also impact the gene-editing efficiency. It is better to select an appropriate transfection method according to the cell type and consider using electroporation for cells that are hard to transfect, such as RPE and Jurkat T cells.78,80
In addition to obtaining sufficient expression levels, the unbiased and extended-expression of PE also affects its ability to write nucleotide alterations into target sites. For example, the inclusion of all the PE3 elements and the selection marker in a single vector has been shown to improve the PE efficiency to ∼95% after nucleofection and antibiotic selection. 67 Furthermore, by using the piggyBac transposon system, the various PE components and the selection marker can be integrated into the host genome. This method has been shown to lead to PE efficacies of up to 100% in HEK293T cells following 3 weeks of antibiotic selection. 77
In addition, fluorescence-activated cell sorting can also enrich the edited cells81,82; for example, a fluorescent PE and enrichment reporter (fluoPEER) has been established by placing a target region (which contains a stop codon or frameshift) between eGFP and mCherry expression cassettes. If the fluoPEER is corrected by the PE machinery, then it leads to the expression of both eGFP and mCherry proteins; otherwise, only eGFP is expressed. 82 When two different pegRNAs (one targeting fluoPEER and the other targeting genomic sites) are simultaneously transfected into cells, sorting the GFP+Cherry+, fluoPEER-enriched population, can enhance the intended genomic editing efficiency with a threefold increase over the completely transfected cell population (GFP+).
Stabilizing the pegRNA structure and hijacking the DNA mismatch system
The PE efficiency is governed by its gene-targeting activity and mutation writing ability. Although the indel frequencies generated by Cas9 nuclease are high at some sites, the PE activity is not always consistent with its nuclease cleavage activity. 83 This indicates that the M-MLV RT, pegRNA extensions, pegRNA scaffold sequences, or cellular context might also contribute to the PE efficacy and specificity.
The codon-optimized M-MLV RT is enhanced for its DNA polymerase activity by templating the nicked 3′ flap, as discussed above. However, its reverse transcription capacity could be enhanced further if an Rad51 DNA-binding domain (which facilitates binding of the pegRNA to the nicked target ssDNA) is placed between the Cas9 H840A nickase and RT domains. Although this new version of PE has a higher prime-editing efficiency than PE2 at endogenous target sites, a major drawback is the increased levels of unintended substitutions and edits (e.g., indels) that occur. 84
Although a high fidelity of gene editing can be achieved by PE2 under the surveillance of a series of DNA/RNA hybridization processes, unintended indels are unavoidably introduced due to pegRNA scaffold insertions or replacements. These are caused by the reverse transcription of 3′-extended pegRNAs into the gRNA scaffold. 28 In the case of PE3 editing, unintended indels do not seem to be directly caused by double nicking per se, but by the combinatory activity of the RT and the pegRNA. 85 To overcome the shortcomings of the PE2 and PE3 platforms, a pair of pegRNAs, which encode the same edits to target both sense and antisense DNA strands, were developed. This new approach was called the homologous 3′ extension-mediated prime editor (HOPE). HOPE shows greatly improved product purity and comparable or higher editing efficiency, when compared with the original PE3 system. 86
Compared with canonical sgRNA, pegRNA contains two additional regions at its 3′-end, an RTT and a PBS. These make the 3′extension more easily exposed in cells, and thus more susceptible to exonucleolytic degradation. The truncated pegRNA might occupy the PE protein and DNA target sites to compete with the function of normal pegRNA. 68 In addition, the PBS region is liable to pair with the spacer sequence, leading to pegRNA circularization and reduced prime editing. 78 Thus, sustaining the structural stability and integrity is crucial for the performance of pegRNA. For this purpose, RTTs starting with a “C” should be avoided as this pairs with G81 in the RNA scaffold, and impairs the gRNA structure, which reduces the editing efficiency 28 ; however, the substitution of A/U base pairs to G/C in the small hairpin of pegRNA scaffolds leads to the formation of apegRNA, which circumvents the disturbance of its secondary structure by the free-swinging 3′ extension.
This results in the intended editing occurring with high efficiency, although there is also a slight increase in the frequency of unintended indels on the target sites. 87 Noncircularizable and non-degradable derivatives of canonical pegRNA can be constructed by fusing different structured RNA motifs to the 3′end of pegRNAs. These motifs include the Csy4 recognition site RNA, 78 G-quadruplexes, 88 pseudoknot RNA motifs (“evopreQ1” or “mpknot”), 68 and a viral exoribonuclease-resistant RNA (xrRNA) (Fig. 3). 89 pegRNA equipped with any of these structured RNA motifs can improve the pan-target average editing efficiency up to 4.5-fold without sacrificing fidelity, when compared with the original PE.
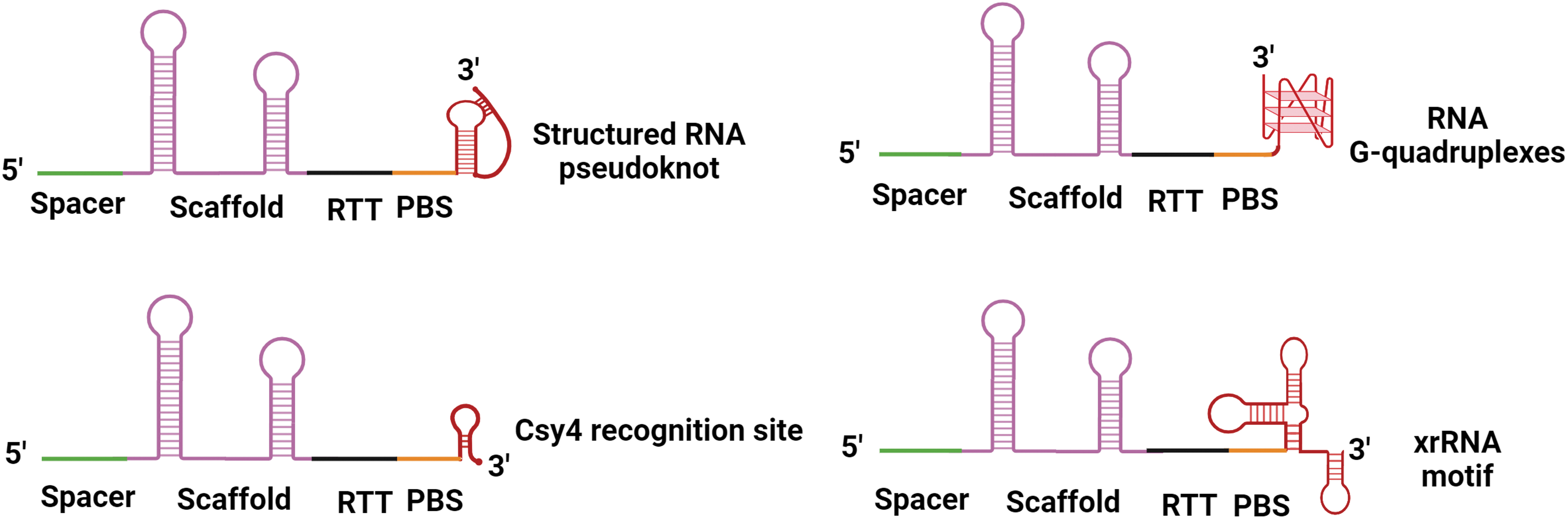
Schematic illustration of a representative pegRNA with 3′ extended modifications. xrRNA, viral exoribonuclease-resistant RNA.
PE has been successfully and widely utilized in a variety of species, including bacteria, yeast, rice, wheat, fruit fly, mouse, and human.28,67,71,90–92 However, by using an additional ngRNA to target the nonedited strand and thus trigger the MMR pathway, PE3 systems do not always show a higher editing frequency than PE2 even in human cells. 28 Moreover, targeting the same pegRNA to the same genomic locus can also lead to different editing efficiencies across different species. This indicates that the genetic background also plays an essential role in determining the capacity of PE.28,40 A transcriptome analysis between PE3-edited and nonedited cell populations found that DNA repair (especially homologous recombination) and cell cycle-related genes are enriched in edited cells. This supports the hypothesis that PE3 induces the MMR pathway to install intended edits into the targeted genomic. 82
Interestingly, through genetic screening, two independent groups unexpectedly discovered that MMR can localize to sites of PE to invade heteroduplex intermediates and promote their reversion to nonedited sequences. They also demonstrated that the rate of MMR toward the desired edit (i.e., with a ligated edited strand) and a nicked nonedited strand after a heteroduplex formation, which represents the most optimal situation for PE3 activity, was low.40,41 Therefore, they proposed that MMR is detrimental to the editing performance of both PE2 and PE3, and therefore, inhibiting this pathway would benefit the prime editing efficacy. In light of this, a dominant-negative form of MLH1 (a key MMR factor) was fused to the original PE systems to develop PE4 (PE2+MLH1dn) and PE5 (PE3+MLH1dn), and both of these were shown to remarkably favor the efficiency and accuracy of various genome engineering techniques. 41
gRNA Design Rules and Web-Based Tools for PE Editing
A conventional pegRNA for PE2 consists of three elements: a protospacer, a PBS, and an RTT. Sometimes, a secondary nick guide sequence is designed to trigger PE3 programming. Therefore, the independent features of these individual components can impact the final editing outcome when they are assembled. Although unexpected base pairs can form during the cross talk among the different elements (just similar to the PBS probably binds with the protospacer sequence, as described above), which disturbs the spatial structure of pegRNA and affects its efficacy, this condition is not easy to accurately assess based on our current knowledge. Thus, in the next section, we simply summarize the essential characteristics of each gRNA unit that influences the eventual editing effects, before illustrating and comparing the different types of software that are available online for pegRNA and ngRNA.
gRNA design rules
The function of the protospacer in pegRNA and ngRNA is to guide nCas9 to the target site and execute single DNA strand cleavage.28,93 Therefore, it is easily understood that the activity of Cas9 nuclease at a given target sequence (which can be predicted by its DeepSpCas9 score) is closely related to PE activity. 94 As the PBS is responsible for pegRNA binding to the nicked strand of the target DNA, this suggests that a high GC content and melting temperature (Tm) can benefit the efficiency of PE2. 83 Indeed, a PBS of 15 or 9 nt is recommended when the GC content is <40% or >60%, respectively. 83 In addition, a PBS Tm ranging from 35°C to 40°C is likely required to achieve efficient editing in human cells. 86
Unlike the PBS, the GC content in the RTT has only a minimal effect on the performance of PE2, although an initial C nucleotide should be avoided as it disrupts the structure of pegRNA. 83 Moreover, it is preferable to use a G or C as the last templated nucleotide when the RTT length is ≤12 or ≥20 nt, respectively. 83 Indels between positions +1 and +6, counting downstream from the pegRNA nicking site (which is at position +1), can disrupt the PAM. This prevents the nCas9 from re-binding and nicking the edited strand before the nonedited strand is repaired, and so it results in higher editing efficiency and lower indel occurrence. 28 If the PAM cannot be disrupted, then including a PAM editing step in addition to the intended editing is recommended.
Finally, including at least five nucleotides of homology downstream of the intended edit in the RTT is expected to achieve efficient editing in human cells.86,95
In general, PE has been found to achieve a higher correcting efficiency for indels than single base substitutions, and so introducing several synonymous mutations in the RTT might result in a high number of intended base substitutions in the target sites (Fig. 4A). 87 Three possibilities might explain this phenomenon. (1) An increasing number of mismatches between the RTT and the target genomic region prevents Cas9 from rebinding and nicking the edited strand before the nonedited strand is repaired. 28 (2) More mismatches in the RTT can easily weaken its Watson–Crick binding to the 3′ end of the genomic nick, which might facilitate the reverse transcription efficiency. 96 (3) MMR was recently found to prevent PE-mediated gene programming, and so, its activity might be inhibited when there are additional benign mutations near to the desired edit. 41

The role of pegRNA architecture on PE performance.
Chen and colleagues systematically analyzed the impact of the number and position of same-sense mutations (SSMs) in the RTT, on the PE editing outcome. 87 They developed spegRNA, which installs no more than four additional base substitutions besides the editing site in the RTT, and found that the spegRNA significantly improved the editing efficiency. This improvement reached a plateau when two additional base substitutions were introduced. 87 Furthermore, after a comprehensive examination of the relationship between the editing efficiency and the position of the SSMs in the RTT, it was suggested that introducing SSMs at no more than five positions (i.e., 1, 5, 6, 2/5, and 3/6, defining the 3′ end of the RTT as position 1) dramatically increased the frequency of the intended base substitution when the target edit was localized to positions 1, 2, or 3 (Fig. 4B). 87
Although the use of spegRNA led to an ∼1.4-fold higher unintended indel frequency, the editing frequency was on average 353-fold higher when compared with the original pegRNA. Moreover, SSMs can also be adopted in the RTT to boost its editing capacity for small insertions or deletions with acceptable off-target effects. 87
The lengths of the PBS and RTT also act as independent parameters to influence the performance of PE. After plotting the average editing efficiencies for each combination of PBS and RTT lengths, a unimodal distribution was shown. 83 PE was optimal when the PBS and RTT lengths were short (e.g., a PBS of 11–13 nt and an RTT of 10–12 nt), and it was less efficient when the PBS and RTT lengths increased. Overall, a PBS of 13 nt and an RTT of 12 nt are recommended for the initial testing of PE2 efficiency, and these should be expanded to 9–15 and 10–15 nt, respectively, in the second round of testing to select the best combination. 83 During this process, it should be noted that consecutive T nucleotides (≥4) should be excluded from the 3′ extension of the pegRNA, as these are read as transcription termination signals to attenuate pegRNA expression.97,98
The distance between the pegRNA nicking sites and edits, between two edits (in the case of serial substitutions), and between two DNA nicks (in the PE3 system), all contribute to the precision and effectiveness of PE. In general, targeted edits with less than six bases downstream from the pegRNA nicking site are easy to edit, 99 and consecutive edits are suggested to not be spaced more than five nucleotides apart in the RTT, to avoid unfavorable PE. When using the PE3-based strategy, two DNA nicks placed close together should be avoided to prevent DSBs and the generation of unwanted indels. Thus, a distance ranging from 50 bp to 100 bp between the nonedited strand nicks and the pegRNA-mediated nick is recommended.28,71 SpCas9 module-based PE makes nontarget-strand DNA nicks at a position three nucleotides from the PAM, which might restrict its effective editing window.
However, a CRISPR-Cas9 ortholog-FnCas9-based PE machinery, using RT linked to an FnCas9 (H969A) nickase module, can create a nick at a site 6 bp upstream of the PAM sequence, and this dramatically expands the PE range (Fig. 4C). 100
As the canonical pegRNA tends to be circularized and degraded among the PBS and RTT regions in living cells, to avoid this, different RNA appendices are suggested to be added to the 3′ end of pegRNAs as described above. In addition, Liu et al and Feng et al independently split the pegRNA into a single sgRNA and a circular RNA (also, respectively, named as petRNA or pRNA by these two groups) that contained an MS2 aptamer at the 5′ or 3′ end of the separate RTT-PBS sequence, and this circular RNA could be recruited to the target site when RT or nCas9 was fused with the MS2 binding protein-MCP (Fig. 4D).101,102 Although petRNA or pRNA exhibited superior stability and comparable accuracy, the editing efficiency was sometimes lower than its pegRNA counterpart at some genomic sites, suggesting that the design of split pegRNA requires further improvements.
Web-based tools for PE editing
Compared with gene knockout and base editing with a conventional gRNA, which is just 20 nt in length,103,104 PE requires a more complex pegRNA, as described above. Although the manual design of a pegRNA is feasible, careful considerations and cross-reference checks of the pegRNA design rules (including protospacer position, favorable PBS and RTT features, and strand orientation) are required. However, this makes the process complicated, error-prone, and limited for large-scale inputs. Fortunately, several web-based tools have been developed (based on common design parameters), to help design appropriate pegRNA and ngRNA candidates, and their efficiency could then be validated according to the protocol provided by David Liu lab. 105
However, it should be noted that PE remains in its infancy but is evolving rapidly and so the current tools are not able to keep up and provide optimal pegRNA designs. However, they still provide several reference sequences that can be checked manually and used for experimental validation and manipulation.
To date, there are at least eight online applications available for PE design. Even though each varies in its design criteria, most include basic functions such as SpCas9 support, custom input, Anzalone's design rules, and on-target or off-target analysis.67,95,98,106–110 After summarizing the design rationale of these online tools, including batch design capability, PAM editing, RNA structure display, oligonucleotides for pegRNA cloning, and Kim's design rules, we find that Easy-Prime, pegIT, and PrimeDesign have slightly stricter criteria. Moreover, PnB Designer and Easy-Prime support pegRNA design based on Kim's design rules, which are the most recent pegRNA design criteria (Table 2).
Summary of prime editing guide RNA online design tools
PAM, protospacer adjacent motif; PE, prime editor; pegRNA, prime editing guide RNA.
On-Target and Off-Target Evaluation Methods for PE
For genome editing to be clinically effective, PE must achieve therapeutically relevant levels of editing at the on-target site with minimal off-target editing. Even though PE has a lower rate of gRNA-dependent off-target editing than Cas9 and no detectable gRNA-independent transcriptome-wide off-target mutations compared with the base editor, random indels still occur.28,42,111 Therefore, it is imperative to evaluate the on- and off-target effects of PE, and indeed both experimental and computational approaches are already established, which can be chosen based on actual situations.
Polymerase chain reaction (PCR) and next-generation sequencing (NGS)-based methods can experimentally examine the frequency of desirable or undesirable edits executed by PE. For the PCR-based approach, target regions amplified by PCR with the appropriate primers can be Sanger sequenced followed by decomposition through EditR, 112 TIDE, 113 or Snapgene software. However, to achieve a high-throughput analysis of the on-target efficiency and unwanted edits, amplicons can be prepared by a two-round PCR strategy. The first round amplifies and captures the target DNA fragments, and the second-round labels a barcode sequence to permit subsequent deep sequencing. The deep-sequencing results can be resolved by several analytic tools, including CRISPResso2, 114 Cas-Analyzer, 115 and PE-analyzer. 108 Off-target sites of pegRNA and ngRNA can be predicted and ranked using several accessible computational programs, including Cas-OFFinder, 116 CRISPR-GE, 117 CCTop, 118 and CRISPOR. 119
Once potential off-target sites are predicted computationally, their actual existence can be validated through NGS sequencing of the off-target site-flanked PCR products. However, it has been demonstrated that many of the off-targets identified in cells were not predicted by the abovementioned analytical tools. This indicates that unwanted edits are substantially underestimated by computational methods and should be monitored through genome-wide sequencing.120–122
To date, several methods for the genome-wide detection of off-target CRISPR cleavage have been established, including Whole Genome Sequencing (WGS), 123 GUIDE-seq, 124 CIRCLE-seq, 125 and CHANGE-seq. 126 Their principles and discrepancies have been reviewed in detail by Atkins et al. 127 It should be noted that apart from WGS, most of the methods were developed primarily to detect genome-wide off-target effects of Cas9 nucleases rather than nickases. However, nickase-based Digenome-seq (nDigenome-seq) was recently established to profile DNA single-strand breaks induced by Cas9 H840A nickase on a genome-wide scale. This method can therefore be specifically used to analyze the accuracy of PE. Consistent with the results of other off-target assessment methods, nDigenome-seq also demonstrates the high fidelity of PE. 66
Application of PE in Biomedical and Translational Research
Due to its ability to precisely generate any moderately sized genetic variations without causing DSBs, PE has exhibited great potential in biomedical studies as well as various therapeutic applications.
Biomedical research
One basic application of PE is to study gene function by introducing point mutations, deletions, and insertions in cell lines.128–132 This was previously achieved using the HDR-based approach or dual sgRNA-based targeted deletion method.133–137 Moreover, PE can integrate up to 10 bp random sequences at target sites by engineering pegRNA to harbor random sequences between the PBS and the homologous arm of the RTT. This newly developed technology, called Random-PE, shows promise for PAM library construction, enhancer screening, and DNA barcoding. 138 In addition, saturation mutagenesis by PE can be used to experimentally analyze the pathogenicity of hundreds of variants in a given endogenous locus.
For example, to investigate the effects of BRCA2 variants on cell proliferation, a pegRNA library that enables the installation of 426 different BRCA2 variants was transfected into BRCA2 haploidized HEK293T cells, and cell populations were sampled at 6 and 14 days post-transfection. Functional or deleterious variants were then identified by calculating the log-fold change of read counts for each pegRNA between the two time points. 139 Finally, PE can also be used to establish animal models of different human diseases or study the pathological function of gene alterations in vivo. The basic procedure involves injecting a mixture of PE2-encoding messenger RNA (mRNA), pegRNA, and ngRNA into zygotes, followed by blastocyst development and genotyping, after which the embryos are transferred to the oviducts of pseudopregnant foster mothers. Through this method, animal models with gene mutations (either deletion or insertion) have been successfully constructed with observed germ line transmission properties and mimetic phenotypes.61,92,140,141
In vivo and ex vivo PE therapy
The discovery of PE with its versatility, high fidelity, and moderate efficiency represents the arrival of next-generation gene therapies for repairing disease-causing mutations in the human genome. To achieve therapeutic editing, delivering PE to target cells can be accomplished either in vivo, by specific delivery of PE to target cells, or ex vivo, by modifying diseased cells and autologously transplanting corrected cells into the body. Generally, there are two main drawbacks to ex vivo therapeutics as follows: (1) isolated diseased cells sometimes have difficulty surviving outside the body; and (2) the reintroduction of cultured cells into the patient often results in poor engraftment, decreasing the success of the therapy.142,143
However, in vivo therapeutics rely on the direct delivery of genome editors to native pathological tissues, which can be applied in diseases where ex vivo manipulations are not possible. PE has already exhibited the therapeutical potential both in vivo and ex vivo to cure human diseases by correcting pathogenic genetic alterations such as gene mutations, deletions, and insertions. The basic steps for evaluating the performance of PE in animal models include guide design, cell lines based on-target and off-target analysis, in vivo validation of target corrections and by-product rates, and physiological consequences interfered by PE in vivo.
Current in vivo PE applications in mouse models are limited to the liver and retina.58,102,144–147 This is because the liver has effective delivery systems,148,149 and the retina has several unique physiological characteristics such as easy accessibility, immune-privileged status, and the presence of tight ocular barriers.150,151
By liver-target delivery, PE has been successfully adopted to treat alpha-1 antitrypsin deficiency (caused by loss-of-function mutations in SERPINA1), 58 hereditary tyrosinemia type 1 (caused by a large-sequence insertion or loss-of-function mutations in FAH),46,145 familial hypercholesterolemia (caused by gain-of-function mutations of PCSK9), 144 and phenylketonuria (caused by loss-of-function mutations of PAH) in mice models 147 ; their disease symptoms could be, respectively, alleviated by PE-mediated A-to-G/C-to-T corrections, small insertions or large-sequence deletions in the corresponding targets, although their correcting efficiency is modest, especially for the large-sequence manipulation with very low editing efficacy (Table 3); the observed symptom relief in the respective disease models is likely that corrected hepatocytes will gain an advantage in growth, and eventually repopulate the liver. 152
The application of prime editor to the correction of pathogenic gene mutations in animal models
AAV, adeno-associated virus; AdV5, adenovirus 5; PE2, second-generation prime editor; sPE, split prime editor.
In addition, the therapeutic potential of PE was also tested in the retina (Table 3). For example, through intravitreal or subretinal injection, PE could achieve an ∼1.8% point mutation rate when, respectively, selecting DNMT1 and ATP78 as the target genes for a proof-of-concept study.145,146 Moreover, Jang et al further validated the feasibility of PE in the retina by using a Leber congenital amaurosis (LCA) model, which possesses a nonsense mutation of the RPE65 (p.R44X); this disease mutation could be corrected with more than 4% prime-editing efficiencies and without detectable mutations, more importantly, the visual function could be then substantially improved in LCA mice. 145 Since even a 1.17% of the wanted edit in a mouse model of a human genetic eye disease could dramtically benefit the retinal function, the level of PE-mediated editing achieved in preclinical studies could make it applicable during the clinical treatment of genetic diseases of eyes. 153
PE2- or PE3-based strategy was applied in the correction of those disease mutations in mice; although the efficiency was overall comparable, PE3 only induced 0.78% indels on average compared with PE2 in one study. 146 In addition, it was reported that the dose and duration of PE treatment in target organs were positively correlated with the final editing rates.58,144,147 Thus, improving PE delivery efficiency and expanding PE acting time are crucial for PE-mediated in vivo therapy.
In addition, PE is also used to restore pathogenic phenotypes through ex vivo approaches (Table 3).154,155 For example, the FAH gene mutation in hepatic progenitors derived from a mouse model of hereditary tyrosinemia type 1 is corrected by PE ex vivo, and then the corrected hepatic progenitors are transplanted into the liver of hereditary tyrosinemia type 1 mice, leading to their extended survival time. 153 Similarly, when primary fibroblasts with COL7A1 mutations derived from patients of recessive dystrophic epidermolysis bullosa (RDEB) were corrected by PE, it was able to rescue the phenotype of RDEB after transplantation of these PE-corrected fibroblasts into immunodeficient mice, which were commonly used as the RDEB model. 154 During ex vivo therapeutics, PE is often delivered into parental cells through a nonviral electroporation method, and corrected cells would be free of PE elements; therefore, when recipients are transplanted with edited autologous cells, lower immune responses will occur in subjects.
Moreover, the selection of gene-corrected cells and non-off-target cells in vitro can increase the efficiency and safety of the ex vivo therapy, but this process might be more cumbersome and costly than in vivo therapy.
In vivo delivery of PE by adeno-associated virus
Compared with Cas9, the larger size of the PE system restricts its efficient in vivo delivery. To address this problem, Böck et al used the human adenovirus 5 (AdV5) vector, which is hepatotropic and could package the full-length PE in a single vector, to deliver PE into mice. 147 Although AdV-mediated PE achieved up to nearly 60% editing rates in the mouse liver, 147 it could induce a potent innate immune response in the body, which is unacceptable in the clinic. A commonly used clinical gene carrier is the adeno-associated virus (AAV) vector due to its superior biosafety, tissue specificities, and low immunogenicity156,157; however, its packaging capacity is only around 4.8 kb, which fails to deliver PE2 (∼6.3 kb) in a single vector.158,159
Various synthetic delivery platforms, such as lipid nanoparticles,160,161 gold nanoparticles,162,163 and Cas-peptide complexes,164,165 have been modified to allow transient delivery of the CRISPR-Cas system into cells without viral components and packaging capacity limitation. However, their synthesis is generally more complex than viral propagation, and they typically have a lower transduction efficiency, when compared with evolved viral coats. To overcome the reduced loading capacity of AAV, a dual-AAV strategy, which involves either trans-splicing AAV vectors (tsAAVs) or using an intein-split system, has been adopted in some preclinical studies.166–168 In these studies, PE was first divided into an N-terminal and C-terminal half, after which subsequent coinfection of these AAV particles was shown to result in the reconstitution of PE elements at the mRNA level (for tsAAV) (Fig. 5A), or the protein level (for the split-intein) (Fig. 5B).

The split-AAV-based strategy to recover the expression of intact or RNaseH domain-deleted PE systems.
It should be noted that the tsAAV strategy seems to be less effective than the single-vector or split-intein AAV systems, and thus might therefore restrict its therapeutic potential.58,145,169 The intein-split-based PE system is also problematic as its large size leaves only a narrow split-site window to fit in the AAV vectors, and so, the optimal split site might be missed (Fig. 5C, D).58,146 In the M-MLV RT moiety, the RNaseH domain (of ∼0.6 kb) was found to be dispensable for RT generation of short products (∼30 bp), and a truncated PE2 (PE2ΔRnH) was observed with comparable editing rates or indel formation compared with PE2.144,147,170 Therefore, deletion of the RNaseH domain enables a 10% reduction in the size of PE2, which provides more opportunities for correct split-site selection (Fig. 5E).
For this reason, several Cas9 split-sites, such as Cas9 E573, Cas9 Q674, and Cas9 K1153, can be selected for dual AAV vector application, as shown in Figure 5F and G, and this has already exhibited robust performance.144,146,147,170 For instance, by splitting Cas9 at E573, the split-PE2ΔRnH can even copy the editing efficiency of PE2ΔRnH up to 92.6% in cell lines, and an in vivo study demonstrated that it could generate an insertion rate of up to 13.5% in the Pcsk9 gene locus. 144 In addition to performing a split in Cas9, a recent study showed that RT elements can engage the RNA–DNA hybrid at the Cas9 nicking site without direct fusion to Cas9. This indicates that Cas9 (∼4.1 kb) and RT (∼2.1 kb) -split PE2, can be packaged separately into an AAV vector (Fig. 5H).
In addition, when untethered nCas9 is cotransfected with RT, it can achieve a similar editing efficiency and specificity compared with full-length PE both in vitro and in vivo. 102 This phenomenon was also similarly observed in Joung's group, and in their recent work, they further split PE2 into nCas9 and RTΔRnH -split PE2-ΔRH, which then demonstrated that split PE2-ΔRH exhibited comparable activity with an intact PE variant. 171 Moreover, they successfully engineered an RT from Eubacterium rectale (Marathon-RT, ∼1.3 kb in size) to achieve acceptable editing performance. Then, a new split PE (sPE) version, which contained nCas9 and Marathon-RT or Marathon-RT ΔRnH, was established. Although Marathon-RT has a smaller size than M-MLV RT, its editing activity appears to be damped, which indicates that the Marathon-RT might require further engineering. 171
Nevertheless, those new versions of the sPE strategy seem more encouraging than the split-intein system, as the latter might accumulate some unassembled by-products that sacrifice its efficiency. Thus, by using the split-vector system, both fragments must simultaneously reach the target site to guarantee a successful outcome.
Outlook
The rapid development of PE technology has greatly improved our ability to make precise changes to the genome of eukaryotic cells. Although the initial PE system had a relatively low editing efficiency compared with cytosine and ABE, which generate ∼50% and ∼40.87–64.22% correction rates, respectively, in their latest variants,22,23 much has been done to evolve PE from its protein and gRNA compositions. For example, by coupling the dominant-negative form of MLH1 to PE3, PE5 can achieve the desired base substitution with ∼40–50% efficiency. 41 In addition, compared with PE3, a combination of PE5 and spegRNA or apegRNA can lead to ∼16.6- or ∼3.2-fold increases, respectively, in the intended base editing frequency. 87 In addition, with the aid of pegRNA pairs, PE is now capable of large-size sequence insertion, inversion, deletion, or chromosomal translocation, although there is still room for improvement.
Nevertheless, this expanded editing versatility paves the way for the treatment of human diseases caused by large deletions, duplications, translocations, or inversions.
Although PE has undergone a remarkable expansion in development in the past 3 years, several bottlenecks remain, which restrict its wider application both in basic biomedical research and clinical translation. For example, PE is reported to induce a lower rate of off-target editing compared with Cas9, but it still occurs especially when secondary nick sgRNAs are used. Although many Cas9 variants exhibit improved specificity, such improvement is built on the sacrifice of its on-target DNA cleavage activities. Recently, a new Cas9 variant-SuperFi-Cas9 was designed, which is reported to achieve wild-type on-target cleavage rates but with higher discrimination against mismatches. 172 Therefore, SuperFi-Cas9 might be adopted in PE to achieve safer genetic alterations but still maintain the original editing efficiency. In addition to the accuracy, the efficacy of PE can be largely controlled by an optimal pegRNA design.
Although many pegRNA design web tools are available, their prediction models are based on out-of-date pegRNA characteristics, and none provides a ranking score to inform users which pegRNAs are highly recommended. Therefore, a more comprehensive and accurate computational model should be established to reduce the labor-intensive pegRNA construction and screening process.
PE has been proven to have high fidelity by genomic and transcriptomic analyses of guide-dependent and guide-independent off-target mutations. However, a recent study found that the RNase H domain in the M-MLV RT moiety can interact with the protein translation termination factor eRF, thereby promoting stop codon read through. 144 Thus, the global protein translation efficiency might be mitigated when intact PE2 is transfected into cells; this possibility can be validated through ribosome profiling assays. 173 By evading MMR, PE4 and PE5 have a dramatically increased editing outcome performance. However, unfortunately, the additional MLH1dn makes their size up to ∼8.7 kb, which substantially limits their AAV package and in vivo delivery. 41
This issue might be addressed in the following three ways. (1) Pretreatment of MMR inhibitors or MLH1 small interfering RNA (siRNA) before PE administration instead of codelivery of MLH1dn. (2) Fusing MLH1dn to a mini Cas protein, such as IscB, which is less than half the size of Cas9. 174 This might enable the installment of the new PE system into a single AAV vector. (3) Several engineered virus-like particles (such as SEND, mLP, and eVLP) have been developed to deliver CRISPR-Cas9 in the form of ribonucleoprotein or mRNA without inducing significant immune responses.175–177 The feasibility of using virus-like systems to deliver the PE platform remains unclear and requires further investigation.
The current PE-mediated direct gene corrections have only been made in the liver and retinal tissues of mouse models, which are established through the installment of a single mutation-single candidate pathogenic gene. Therefore, future studies should broaden its application to other organs or multigene genetically caused disease animal models; the former might be addressed by developing new gene carriers with enhanced infectivity and tissue-specific tropism, while the latter is likely to be resolved by a recently established PE3 gRNA array system or p2PE3 system, which could both achieve up to 3-loci multiplex prime editing at the same time.79,178 Although it is still too early to justify the therapeutic potential of PE, this technique has a promising outlook due to the expanding arsenal of techniques and delivery systems that are being developed.
Footnotes
Acknowledgment
The illustrations were created with BioRender.com
Authors' Contributions
D.W. and J.Z. conceptualized and wrote the article; D.W., X.F., and M.L. prepared the illustrations; and T.L., P.L., Y.L., G.W., and J.H. reviewed the article.
Author Disclosure Statement
All the authors declare no potential conflicts of interest.
Funding Information
This work is supported by the National Key Research and Development Program of China (2017YFC1309800), the National Natural Science Foundation of China (82130025 and 32200557), and the “Outstanding University Driven by Talents” Program and Academic Promotion Program of Shandong First Medical University (2019LJ007).