
Abstract
Microinjected transgenes, both large and small, are known to insert randomly into the mouse genome. Traditional methods of mapping a transgene are challenging, thus complicating breeding strategies and accurate interpretation of phenotypes, particularly when a transgene disrupts critical coding or noncoding sequences. As the vast majority of transgenic mouse lines remain unmapped, we developed CRISPR-Cas9 Long-Read Sequencing (CRISPR-LRS) to ascertain transgene integration loci. This novel approach mapped a wide size range of transgenes and uncovered more complex transgene-induced host genome re-arrangements than previously appreciated. CRISPR-LRS offers a facile, informative approach to establish robust breeding practices and will enable researchers to study a gene without confounding genetic issues. Finally, CRISPR-LRS will find utility in rapidly and accurately interrogating gene/genome editing fidelity in experimental and clinical settings.
Introduction
Despite the routine use of transgenic mice, >90% of transgenic alleles in the Mouse Genome Database have yet to be mapped1,2 even though it is broadly accepted that random integration of a transgene can disrupt coding exons and functional noncoding sequences (e.g., enhancer, long noncoding RNA [lncRNA]), thus complicating correct interpretation of phenotypic data from transgenic mouse studies. Fluorescence in situ hybridization can map a transgene to a chromosome, but lacks nucleotide resolution. 3 Hybridization-microarray, targeted locus amplification (TLA), and linear amplification-mediated polymerase chain reaction (PCR) rely on short-read sequencing and cannot resolve complex genome inversions/deletions.4–7 Inverse PCR (iPCR) has a high failure rate due to the concatemer nature of most transgenes.8,9 Recently, whole genome sequencing (WGS) with long-read sequencing platforms, PacBio or Oxford Nanopore Technologies (ONT), have been used to map transgenes.2,10 But even ∼3 × sequence coverage of the genome may not yield a transgene integration locus (i.e., transgene and mouse genome breakpoint). The challenge in mapping a transgene lies within the unpredictable nature of the transgene as its integration can result in a myriad of potentially harmful host genome perturbations.1,2,5,10–12
In this study, we propose a more targeted/enrichment and non-PCR based approach to map transgenes by combining the substantial genome sequence coverage of the ONT sequencing platform13,14 with the RNA programmable CRISPR-Cas9 system. 15 Previous studies have combined these two technologies to interrogate specific genomic loci for a number of goals,16–23 but at the time of this writing, none have used this approach to map a transgene in an animal model.
For proof-of-principle, we chose a wide range of sized transgenes, 1 to >200 kb, to map to test the efficacy of CRISPR-Cas9 Long-Read Sequencing (CRISPR-LRS). To this end, both large (Bacterial Artificial Chromosomes [BACs]) and small (Cre-driver and ECFP-reporter) transgenes were efficiently mapped with this approach. CRISPR-LRS represents a facile tool for mapping a transgene, thus informing investigators as to best breeding practices and identifying any potentially confounding genetic element(s) within their transgenic mouse lines. Further, we imagine CRISPR-LRS will find utility in assessing the fidelity of gene/genome edits in all life forms.
Methods
Transgenic mice
Transgenes, both large and small, were mapped for five different mouse lines (Supplementary Table S1). The RP11-744N12 BAC with human SENCR, Sm22-Cre, and Thy1-ECFP-VAMP2 mouse lines were reported previously24–26 and CTD-2518N7 and RP11-997L11 BAC transgenic lines were generated by Cyagen (www.cyagen.com) using strain C57BL/6J. With the exception of Thy1-ECFP-VAMP2, all mice were maintained on the C57BL/6J background with repeated back-crossing and refreshing breeders every five generations to mitigate genetic drift.
Breeding details for Thy1-ECFP-VAMP2 line can be found in Liu et al. 26 Mouse experiments were approved by Medical College of Georgia at Augusta University Institutional Animal Care and Use Committee (Approval Nos. 2019-1000 and 2019-0999).
Long-read library preparation (CRISPR-LRS and WGS)
Long-read libraries used the Targeted-CRISPR-LRS or the Enrichment-CRISPR-LRS approach (see the Results section for details). For the BAC lines and the Sm22-Cre line, liver genomic DNA (gDNA) was isolated using Qiagen DNeasy Blood & Tissue Kit (cat#69504) following the manufacturer's instructions (www.qiagen.com). For Thy1-ECFP-VAMP2 line, liver gDNA was isolated using NEB Monarch HMW DNA kit (cat#T3060) following the manufacturer's instructions (www.neb.com).
To limit shearing of gDNA, wide bore pipette tips were used. Both Targeted-CRISPR-LRS and Enrichment-CRISPR-LRS libraries used 5 μg of liver gDNA as input and were prepared following the manufacturer's instructions with the long fragment buffer option (SQK-CS9109) (www.nanoporetech.com). This ONT kit uses a two-part guide RNA composed of CRISPR RNAs (crRNAs) and trans-activating CRISPR RNA (tracrRNA) to program the ribonucleoprotein (RNP).
crRNAs were designed using CHOPCHOP 27 with default parameters (Supplementary Table S2) (https://chopchop.cbu.uib.no). All crRNAs, tracrRNA, and HiFi Cas9 were ordered from IDT (www.idt.dna.com) following ONT suggestions.
For WGS with ultra-long-reads, liver gDNA from the Sm22-Cre line was isolated using circulomics kits (i) nanobind tissue big DNA kit (cat#NB-900-701-01), (ii) nanobind ul library prep kit (cat#NB-900-601-01) and (iii) UHMW DNA aux kit (cat#NB-900-101-01) (www.circulomics.com). Ultra-high molecular weight (uHMW) gDNA was used as input for Ultra-Long DNA libraries, which were prepared with the Ultra-Long DNA Sequencing Kit (SQK-ULK001) following manufacturer's instructions (www.nanoporetech.com).
Nanopore sequencing and data analysis
CRISPR-LRS libraries (SQK-CS9109) and WGS with ultra-long-read libraries (SQK-ULK001) were run on R9.4.1 flow cells on either the minION Mk 1B or the GridION platform following the manufacturer's instructions (www.nanoporetech.com) (see Supplementary Table S3 for library metrics). Reads were converted from fast5 to fastq with guppy (v4.2.2) on MinKNOW (v20.10.3) MinKNOW Core (v4.1.2) with fast base-calling option for the base-call model and with a minimum Q-score of 7 option for the read filtering option.
To analyze long-read sequencing results, guppy base-called fastq files were imported into Qiagen CLC Genomics Workbench (www.qiagen.com). Reference sequences specific to the transgene to be mapped were obtained from NCBI nucleotide database (www.ncbi.nlm.nih.gov/nucleotide/). Fastq files were aligned to the reference sequence with the Qiagen CLC Genomics Workbench Long-Read Support (beta) plugin with default parameters (www.digitalinsights.qiagen.com).
The plugin utilizes components of open-source tool minimap2. 28 Aligned informative long-reads were extracted and manually queried against NCBI nr/nt and refseq genome databases (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and UCSC genome browser with BLAT tool (https://genome.ucsc.edu). Graphical output obtained from CLC Genomics Workbench and GraphPad (www.graphpad.com) were amended with Adobe Illustrator (www.adobe.com) (Adobe Systems, San Jose, CA) for illustration purposes.
RNA-Seq
Total RNA was isolated from the spleen of RP11-744N12 BAC (tg/tg) and (+/+) mice using Direct-zol RNA MiniPrep kit (cat#ZR2052) from Zymo Research following the manufacturer's instructions (www.zymoresearch.com). RNA-Seq Illumina libraries were prepared and run at University of Rochester Genomics Research Center using TruSeq Stranded kits and run on a NovaSeq 6000 instrument (www.urmc.rochester.edu). RNA-Seq analysis was done with Qiagen CLC Genomics Workbench (www.digitalinsights.qiagen.com) using default parameters.
Genotyping and transgene copy number analysis
For genotyping, gDNA was extracted from ear punches using Qiagen DNeasy Blood & Tissue Kit (cat#69504) following manufacturer's instructions (www.qiagen.com). Progeny of (+/tg) × (+/tg) heterozygous crosses were genotyped based on the transgene and mouse genome breakpoints mapped by CRISPR-LRS. PCR conditions: step 1, 95°C for 3 min; step 2, 95°C for 30 s, 58°C for 30 s, 72°C for 1 min for 35 cycles; step 3, 72°C for 10 min. Amplicons for (i) transgene and mouse genome breakpoint queries and (ii) genotyping were validated by Sanger sequencing (Supplementary Table S2).
For transgene copy number quantification, 50 ng of gDNA was used as input. For the BAC mouse lines, two primer sets to the chloramphenicol resistance gene served as a proxy for the BAC transgene. Values were normalized to an internal control locus (Supplementary Table S2).1,2,10 For the Cre-driver mouse line, two primer sets to Cre were normalized to the internal control locus (Supplementary Table S2). For the Thy1-ECFP-VAMP2 mouse line, a primer set to ECFP was normalized to the internal control locus (Supplementary Table S2). The Itga8-CreERT2 mouse with one copy of Cre 29 served as a calibrator for one copy of Cre. Real time quantitative PCR (qPCR) conditions: step1, 95°C for 3 min; step 2, 95°C for 30 s, 60°C for 30 s, 72°C for 30 s for 40 cycles; melt curve analysis.
Data availability
Next Generation Sequencing libraries of CRISPR-LRS, WGS with ultra-long-reads, and RNA-Seq can be found under BioProject number PRJNA759232 at NCBI SRA database (www.ncbi.nlm.nih.gov/sra).
Results
Overview of CRISPR-LRS
As transgene integration can be accompanied by complex host genome rearrangements,1,2,5,10–12 a long-read sequencing platform is key to navigate through these complicated genomic sequence perturbations to find transgene and mouse genome breakpoints. The mechanism of CRISPR-LRS is straightforward and highly effective. To specifically target the transgene over other regions of the genome, the RNP complex is programmed with a Cas9 enzyme loaded with a two-component guide RNA (crRNA and tracrRNA).
This programmed RNP will bind and cleave within the transgene to make a break within the host genome. This RNP-breakage creates a free-end which is then ligated with the ONT adapter to form a duplex. This duplex, of (i) ONT adapter and (ii) RNP-cleaved genomic sequence of variable length, is then shuttled to the ONT sequencing pore to generate the long-read sequence data. The length of these long-reads is only limited by the length of the RNP-cleaved genomic sequence. Our data found long-read lengths with a range of 1000–200,000 bp, substantially longer than short-reads of 150 bp generated with standard PCR-based platforms (e.g., Illumina). Long-reads are then queried to find transgene and mouse genome breakpoint(s) (see the Methods section for details) (Fig. 1A).
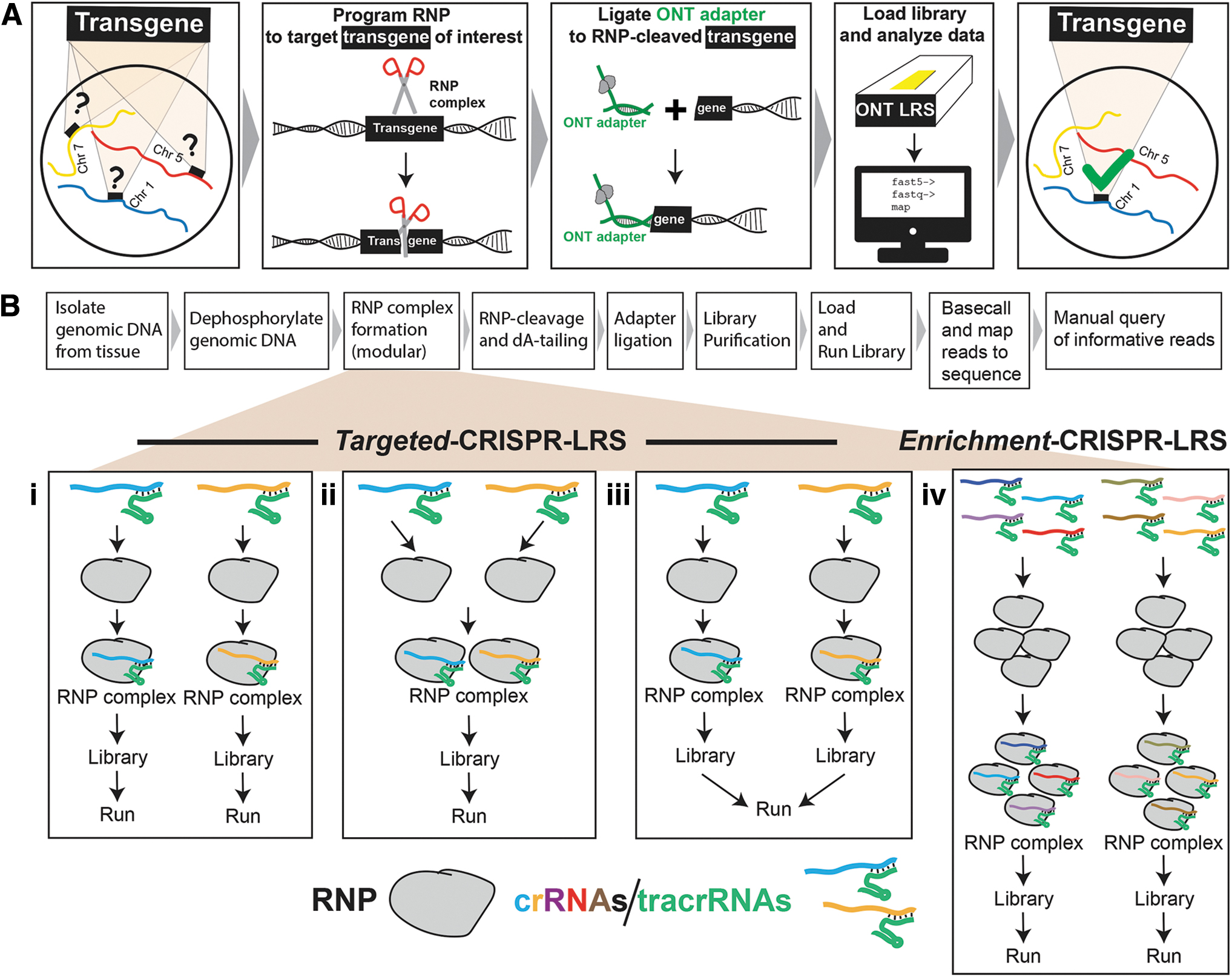
Mechanism and library preparation flow chart for CRISPR-LRS.
CRISPR-LRS can be designed (i) to target a genomic locus (Targeted-CRISPR-LRS) or (ii) to enrich for a specific genomic region (Enrichment-CRISPR-LRS) (Fig. 1B). For Targeted-CRISPR-LRS, 5′ and 3′ ends of the transgene were targeted with RNPs loaded with either one or multiple crRNAs (Fig. 1Bi–iii). For Enrichment-CRISPR-LRS, tandem crRNAs targeted up- and down-stream of the specific genomic region of interest and all crRNAs were loaded into one RNP (Fig. 1Biv).17,23
For the five transgenic mouse lines in this study, eight CRISPR-LRS libraries (Targeted or Enrichment) were sequenced on the minION or GridION platform for a total of ∼2.8 Gb at ∼500,000 reads (Supplementary Tables S2 and S3). Reads were mapped to their corresponding reference sequence with a range of 0.02–1.20% (Supplementary Table S3) and will be called informative long-reads for the remainder of the report.
CRISPR-LRS mapping of large transgenes
A mouse line carrying the RP11-744N12 BAC was generated to study human-specific lncRNA, SENCR. 24 To map the integration locus for the 217 kb BAC, two Targeted-CRISPR-LRS libraries were made to target at the 5 kb region (bottom strand, blue arrow) or at the 3 kb region (top strand, yellow arrow) of the BAC transgene (Figs. 2Ai and 1Bi, and Supplementary Table S2). Both libraries yielded 0.03% of reads (0.9–25 kb) mapped to the reference sequence (Fig. 2Ai and Supplementary Table S3). Notably, informative long-reads found the BAC transgene to be flanked with pBACe3.6 cloning vector sequence with ∼1 or ∼7 kb at the 5′ and 3′ ends of the transgene, respectively (Fig. 2Ai). Informative long-reads >6 or >10 kb for 5′and 3′ ends, respectively, found the RP11-744N12 BAC integrated seamlessly into the first intron of Egflam at 7,344,678 bp on Chr15 (GRCm38/mm10) without any genomic scars (Fig. 2Ai). Of note, seamless integration of a transgene without collateral damage (i.e., no deletions or inversions of the mouse genome), is unusual as genome perturbations usually accompany transgene insertions.1,2,5,10–12

CRISPR-LRS mapping of large transgene RP11-744N12 BAC to chromosome 15 in mouse genome.
As only a few long-reads supported the RP11-744N12 BAC integration locus, we next used Enrichment-CRISPR-LRS to further test the accuracy of our approach. Here RNPs were loaded with crRNAs to enrich for the genomic region containing (i) mouse Chr15, (ii) pBACe3.6 cloning vector, and (iii) RP11-744N12 (Figs. 2Aii and 1Biv). With 5- and 12-fold enrichment of informative reads at the 5′- and 3′-terminal ends, respectively (compare Fig. 2Aii to Ai), Enrichment-CRISPR-LRS validated the Targeted-CRISPR-LRS transgene mapping data.
Sanger sequencing further confirmed the seamless integration of the RP11-744N12 BAC to the first intron of Egflam on mouse Chr15 (Fig. 2Aiii and Supplementary Table S2). Genotyping pups from a heterozygous (+/tg) intercross with primers spanning (i) transgene and mouse Chr15 breakpoint and (ii) wild type mouse Chr15 sequence (Fig. 2B), revealed near Mendelian ratios with 9/41 (22%) wild type (+/+), 17/41 (41%) heterozygous (+/tg), and 15/41 (37%) homozygous (tg/tg) mice.
While no overt pathology was observed with RP11-744N12 (tg/tg) mice, it is quite probable that integration of the 217 kb BAC transgene would compromise expression of the host gene, Egflam. Indeed, RNA-seq of spleen tissue demonstrated lower expression of Egflam in RP11-744N12 (tg/tg) mice over wild type (+/+) mice (Fig. 3). Specifically, RP11-744N12 (tg/tg) mice were nearly null for expression of Egflam up to exon 18, whereas exons 19–23 showed no difference across mutant and wild type, suggestive of an internal promoter for this gene (Fig. 3). Based on these results, expression of the host gene, Egflam, was compromised, thus representing a confounding genetic variable that would need to be taken into account when using this line.
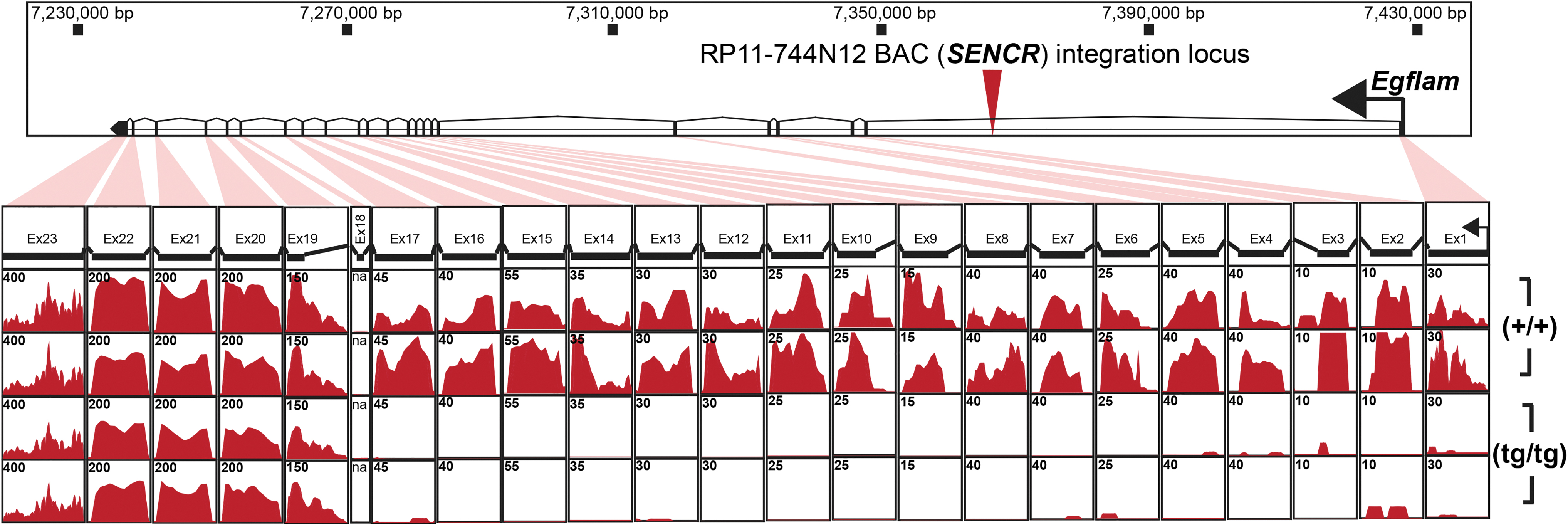
Effect of RP11-744N12 BAC integration on host gene expression. Expression of Egflam, host gene for the BAC, was assessed by RNA-Seq from spleen of (tg/tg) and (+/+) mice. Histograms for read abundance per exon illustrate RP11-744N12 BAC (tg/tg) mice with marked reduction in expression of Egflam compared to (+/+) mice in exons 1–18.
Mouse BAC lines CTD-2518N7 and RP11-997L11 were generated to perform BAC-editing experiments. Such genome editing studies are best carried out with a single copy of the BAC transgene where precision editing is favored over more complex editing outcomes that accompany lines with multiple transgene copies. Targeted-CRISPR-LRS was used to map these BAC lines. Head to tail tandem integration was found for both lines as informative long-reads contained the BAC-cloning vector flanked with 5′ and 3′ ends of the BAC transgenes (Supplementary Fig. S1A, B).
Even though CRISPR-LRS did not map these transgenes, the results indicated each of the BAC lines was unsuitable for further editing experiments, thus saving valuable time, effort, and resources.
As all of the BAC transgenes were flanked with BAC-cloning vector sequence (Fig. 2 and Supplementary Fig. S1), transgene copy number could be determined by quantifying a common gene among BAC-vectors, the chloramphenicol resistance gene. qPCR, routinely used to quantify transgene copy number,1,2,5,10 found RP11744N12 (+/tg) and (tg/tg) mice with one and two transgene copies, respectively (Supplementary Fig. S2A) and found approximately three transgene copies for the tandem integrated BAC (+/tg) mouse lines (Supplementary Fig. S2A). qPCR data agreed with CRISPR-LRS mapping data (compare Supplementary Fig. S2A to Fig. 2).
CRISPR-LRS mapping of small transgenes
Small transgenes are a challenge to map as they can integrate as concatemers (in some cases >50 copies), resulting in complex local re-arrangements of the host genome.1,2,5,10–12 While CRISPR-LRS was able to map large transgenes (BACs), we next tested if it could handle the challenge of mapping smaller transgenes <1 kb.
Targeted-CRISPR-LRS was used to map Sm22-Cre, a mouse line used to excise floxed DNA sequences in early embryonic heart and smooth muscle cell-containing tissues. 25 To map larger transgenes (e.g., BACs), crRNAs were programmed to target at least 2 kb from the terminal ends of the transgene (Figs. 2 and 1Bi, ii and Supplementary Fig. S1), but with the ∼1 kb Cre Recombinase gene, a different mapping approach was used.
Here, two independent Targeted-CRISPR-LRS libraries were made to target at 0.8 kb (bottom strand, blue arrow) and at 0.5 kb (top strand, yellow arrow) of the transgene as a means to overlap the transgene (Figs. 4Ai and 1Biii and Supplementary Table S2). The two independent library preps were then combined to run on a single flow cell to obtain a single library run (Fig. 1Biii).
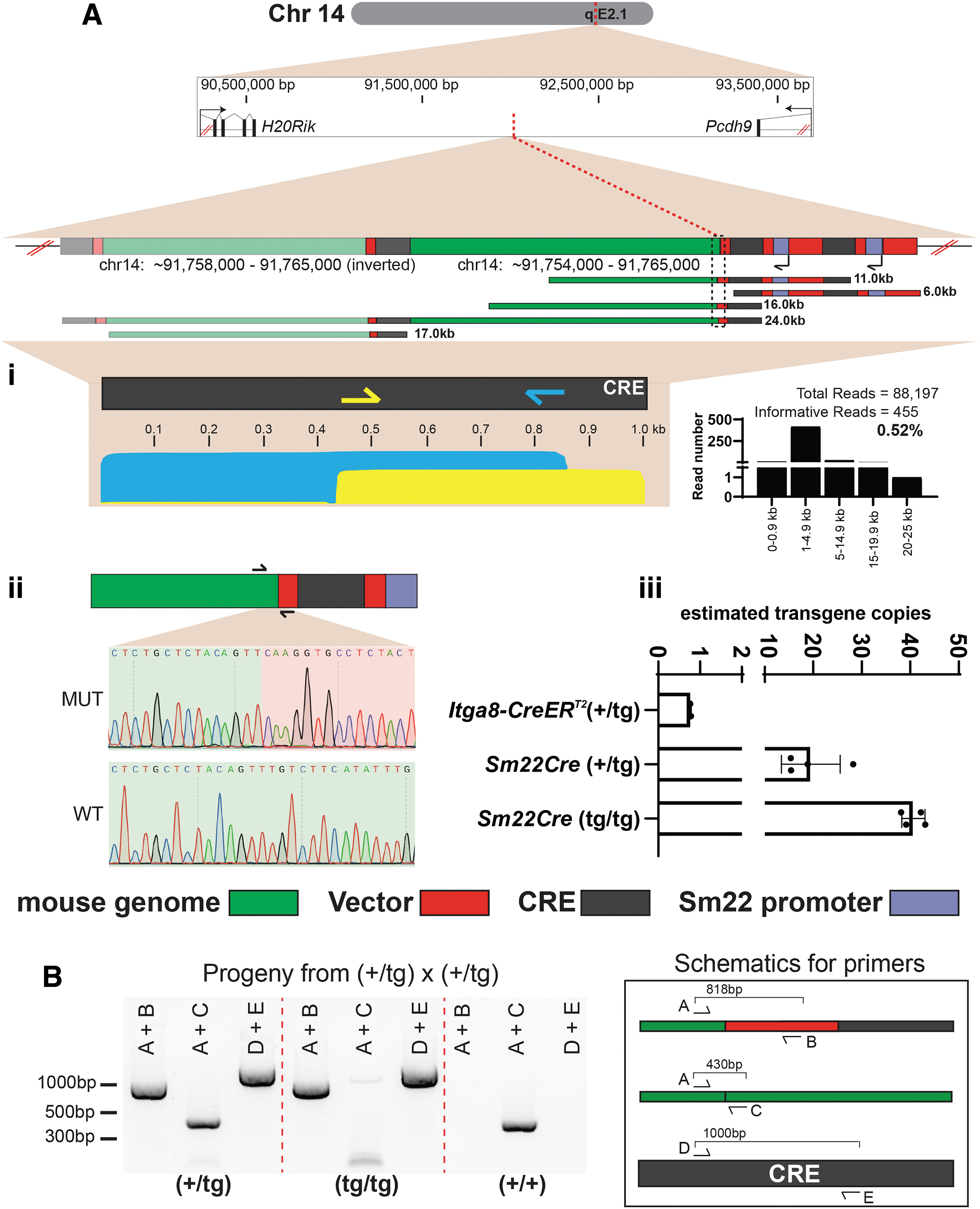
CRISPR-LRS mapping of small transgene Sm22Cre-driver to chromosome 14 in mouse genome.
At 0.52%, the Sm22-Cre library (Fig. 4Ai) contained more informative long-reads compared with the BAC line libraries (Fig. 2 and Supplementary Fig. S1, and Supplementary Table S3). Manual interrogation and alignment of >6 kb informative long-reads elucidated a mini-tiled Cre transgene integration map consisting of a concatemer of four Cre-transgenes within a gene desert followed by re-arrangement, duplication and inversion of mouse Chr14 (Fig. 4Ai).
A transgene and mouse genome breakpoint was identified at 91,527,881 bp on mouse Chr14 (GRCm38/mm10) with sequence support from multiple informative long-reads (Fig. 4Ai, dashed black line box). Sanger sequencing verified this transgene and mouse genome breakpoint (Fig. 4Aii and Supplementary Table S2). Genotyping pups from a (+/tg) × (+/tg) heterozygous intercross with primers spanning (i) the breakpoint of Cre and mouse Chr14 and (ii) wild type mouse Chr14 sequence (Fig. 4B) revealed near Mendelian ratios with 8/41 (19%) wild type (+/+), 20/41 (49%) heterozygous (+/tg), and 13/41 (32%) homozygous (tg/tg) mice. Similar to the RP11-744N12 BAC mouse, no overt pathology was observed in homozygous Sm22-Cre mice.
Targeted-CRISPR-LRS found 4 copies of the Sm22-Cre transgene (Fig. 4Ai), but qPCR determined ∼20 and ∼40 copies for (+/tg) and (tg/tg) pups, respectively (Fig. 4Aiii and Supplementary Fig. S2B), indicating a substantial number of the Cre-transgenes were not mapped. To address this gap, we used a broad sequencing approach using WGS with ultra-long-reads (see Methods section for details).
First, to obtain ultra-long stretches of the genome to sequence, uHMW-gDNA was isolated using a disc-based approach over conventional column-based methods, which can shear gDNA. Second, to obtain greater genome sequence depth, uHMW-gDNA WGS libraries were run on a GridION over the minION. The uHMW-gDNA WGS library yielded 4.3 Gb of genomic sequence with an average read length of ∼50 kb and some informative long-reads >200 kb (Fig. 5 and Supplementary Table S3).
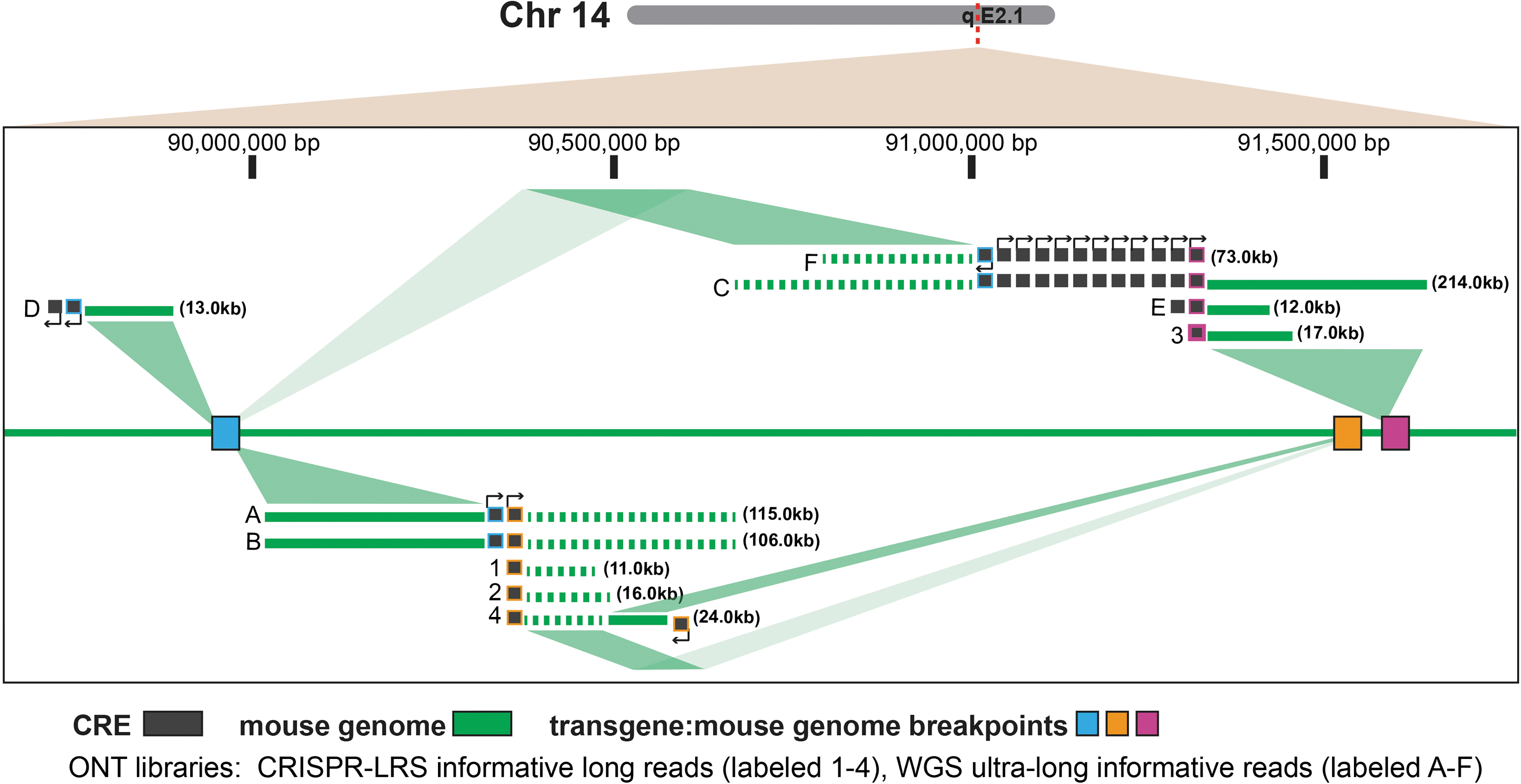
Map of the complex local genome re-arrangements of mouse genome at the Sm22Cre integration locus. Informative reads from ONT libraries of (i) CRISPR-LRS (labeled 1–4) and (ii) WGS with ultra-long-reads (labeled A–F) defined three transgene and mouse genome breakpoints which spans ∼90,000,000 to 91,500,000 bp on mouse Chr14 (GRCm38/mm10). Breakpoints (designated as blue, orange, and pink boxes) are supported by long-reads of solid and dashed lines, which represent top and bottom strands of the genome, respectively. The Sm22Cre integration locus map illustrates ∼17 copies of Cre (designated as black boxes) where orientation of each transgene is designated by a promoter arrow. ONT, Oxford Nanopore Technologies; WGS, whole genome sequencing.
Even with a finite number of informative long-reads (0.00003%, Supplementary Table S3), this sequencing approach identified the same transgene and mouse genome breakpoint that Targeted-CRISPR-LRS defined (Figs. 4A and 5) as well as two additional transgene and mouse genome breakpoints, with the Sm22-Cre integration locus spanning from ∼90,000,000 to 91,500,000 bp on mouse Chr14 (GRCm38/mm10) (Fig. 5).
Notably, all three transgene and mouse genome breakpoints contained concatemers flanked by top- and bottom-strands of mouse Chr14 sequence (Fig. 5). Lastly and most impressively, the uHMW-gDNA WGS data found 17 copies (Fig. 5), similar to ∼20 copies of the Sm22-Cre transgene determined by qPCR (Fig. 4Aiii and Supplementary Fig. S2B). The uHMW-gDNA WGS approach yielded a broader view of the complex local genomic re-arrangements, which accompanied integration of the numerous copies of the Sm22-Cre transgene (Fig. 5).
But, even though uHMW-gDNA WGS data did illustrate a more complex Sm22-Cre integration locus (Fig. 5), Targeted-CRISPR-LRS did define a breakpoint between the Cre-transgene and mouse Chr14 genome (Fig. 4A) that was successfully used for genotyping and basic colony maintenance (Fig. 4B).
Targeted-CRISPR-LRS was used to map another small transgene, a reporter line, Thy1-ECFP-VAMP2, used to study protein aggregates with a neuron-specific promoter. 26 To improve read length and overall sequence depth, HMW gDNA was isolated by glass beads and libraries were run on a GridION. To map the 0.7 kb ECFP transgene, two independent Targeted-CRISPR-LRS libraries were made to target at 0.6 kb (bottom strand, blue arrow) and at 0.35 kb (top strand, yellow arrow) of the transgene as a means to overlap the transgene (Figs. 6ii and 1Biii and Supplementary Table S2). Similar to the Sm22-Cre mapping above (Fig. 4), the two independent libraries were then combined onto one flow cell to obtain a single library run (Fig. 1Biii). Across all CRISPR-LRS libraries, the ECFP-reporter library contained the highest number of informative reads (1.2%) and greatest average read length (23.3 kb) (Fig. 6ii and Supplementary Table S3). Manual interrogation and alignment of informative reads >30 kb found three breakpoints of transgene and mouse genome for four copies of the Thy1-ECFP-VAMP2 transgene along a gene desert (based on RefSeq gene models) at ∼23,705,000–23,870,000 bp on mouse Chr15 (GRCm38/mm10) (Fig. 6i).
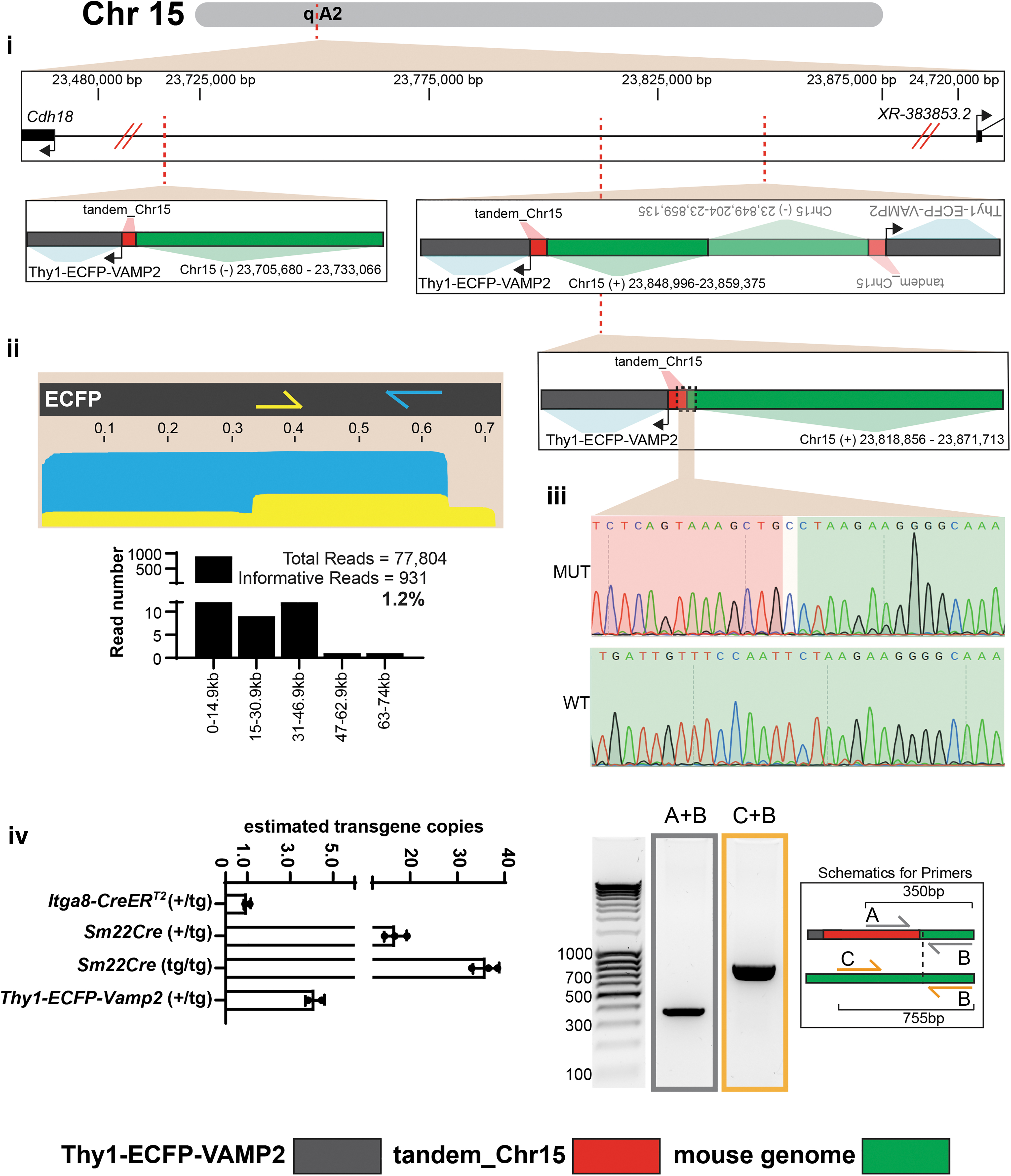
CRISPR-LRS mapping of small transgene Thy1-ECFP-VAMP2 to chromosome 15 in mouse genome.
Transgene and mouse genome breakpoint at 23,818,856 bp on mouse Chr15 contained the best support from multiple informative long-reads (Fig. 6i) and was verified by PCR and Sanger sequencing (Fig. 6iii). Lastly, qPCR determined approximately four copies of the transgene, in agreement with the Targeted-CRISPR-LRS mapping data (Fig. 6iv). Curiously, each of the four transgenes were preceded by a 1.5 kb section of top- and bottom-strand of Chr15 fused together in tandem (Fig. 6i).
Discussion
While a transgene typically integrates to a single locus,3,30 the integration site is highly variable with multiple copies of the transgene as well as complex local genome re-arrangements.1,2,5,10–12 These changes to the host genome can result in insertional mutagenesis, which can alter expression of endogenous gene(s) in an unpredictable manner resulting in confounding genetics that could mask, inhibit, or enhance transgene phenotype(s).1,2,5,10–12 Cases of such insertional perturbations include: (i) >20 unmapped Cre-driver mouse lines with a phenotype in transgene isolation, 31 (ii) three mapped transgenes that act as an “enhancer trap,” 11 (iii) ∼50% of mapped recombinase-transgenes with integration loci that disrupt endogenous genes, 1 and (iv) previously reported phenotypes later determined to be better explained by effects of “insertional mutagenesis” over effects of the transgene.11,32,33
It is important to note that transgene-induced alterations to the host genome can span multiple kilobases with transgene copy number exceeding >50 copies,2,5 a major challenge for short-read sequencing platforms which struggle with sequencing through and mapping repetitive genomic sequences. 34 In contrast, the long-read sequencing platform can sequence through and map repetitive genomic regions17,35 as demonstrated by its use to fill sequence gaps of human chromosome8.36,37
To test the efficacy of CRISPR-LRS, transgenes ranging from 1 to >200 kb were selected to be mapped. Here, mapping data found massive variation across integration loci for the five lines consisting of one or more of the following variables: (i) cloning-vector sequence of various length (0.5–13.0 kb) flanking the transgene, (ii) complex re-arrangements of the host genome, and (iii) numerous copies of the transgene. For small transgenes, CRISPR-LRS determined copy number, resolved complex local genome re-arrangements at the integration locus, and defined transgene and mouse genome breakpoints for a Cre- and a reporter-line. For big transgenes, CRISPR-LRS determined head to tail integrations for two BAC alleles, designating them to be limited for purposes of BAC editing experiments. For the BAC line with the human lncRNA SENCR, 24 CRISPR-LRS determined a scar-less integration in the first intron of Egflam, where follow up RNA-seq analysis found a severe reduction in Egflam expression in SENCR (tg/tg) mice. This last data point serves as another example of the effects of “insertional mutagenesis” and demonstrates the need to account for this phenomenon1,11 as well as to possibly reassess phenotypes from studies that used lines without a mapped transgene.
Of the available strategies to map transgenes, iPCR,8,9 WGS2,10 and TLA1,5 have the most traction. However, iPCR is limited to mapping low copy number transgenes,5,8,9,11 whereas CRISPR-LRS mapped multiple complex concatemers with head to tail or head to head orientation (Figs. 4–6 and Supplementary Fig. S1). Further, iPCR cannot illustrate any structural changes at the transgene integration site,5,8,9,11 while CRISPR-LRS illustrated a greater breadth of genome re-arrangements than was previously appreciated (Figs. 4–6 and Supplementary Fig. S1). WGS is not a targeted approach to map a transgene as the majority of reads are uninformative.2,10 Here with only ∼2.8 Gb of sequence data, CRISPR-LRS (i) determined five mouse lines to have single or multiple transgene copies, and (ii) established transgene and mouse genome breakpoints for three mouse lines. For TLA, this approach can be technically challenging for most labs as (i) it has numerous steps; crosslinking, fragmentation, religation, and amplification, and (ii) analysis can require extensive computational expertise due to the nature of these fragmented sequence libraries.1,5 CRISPR-LRS (i) has less steps (i.e., Cas9 cleavage and adaptor ligation), and (ii) only requires mapping long-reads with an open source tool, minimap2 (Fig. 1). 28
Even though CRISPR-LRS proved effective at mapping transgenes with complex integration loci, improvements in certain areas will make it more approachable and useful for the mouse community. One area to improve is increasing the sequence coverage and depth for breakpoint(s) of the transgene and host genome. It should be noted that low coverage is to be expected as CRISPR-LRS sequences the native genome, not PCR-amplified libraries. To this point, we have increased (i) sequence-coverage with greater length of long-reads by isolating longer stretches of HMW gDNA with glass beads over a column and (ii) sequence-depth with the use of the higher fidelity GridION (see Methods section for details) (compare Fig. 6 to Figs. 2 and 4 and Supplementary Fig. S1).
While fruitful, continued efforts to strengthen this area will pay dividends for future efforts to map transgene integration loci with even greater complexity than here, and to scale up CRISPR-LRS for mapping multiple transgenes in one library. Another area to improve is optimizing parameters of gDNA isolation to obtain sufficient material for library preparation in a nonlethal manner. In this study, mice were culled to obtain enough starting material for library construction.
While our method proved effective, this approach to map transgenes is not ideal for screening founder mice. Recently, a transgene mapping approach based on Circle-Seq called CRISPR-KRISPR was reported to use low amounts of gDNA as input for library construction. 38 This short-read sequencing platform represents a complementary mapping approach to CRISPR-LRS with the added advantage of interrogating off-targeting and random insertional events following CRISPR editing.
To start to map a transgene, we suggest using Targeted-CRISPR-LRS to define breakpoint(s) and/or tandem integration for a transgene (Figs. 2, 4 and 6 and Supplementary Fig. S1). If sequence coverage of the mapped breakpoint(s) is low, then Enrichment-CRISPR-LRS should be used to obtain greater sequence coverage for more confidence in defining the breakpoint for use in basic mouse line maintenance (Fig. 2Ai, Aii). If qPCR reveals a high number of copies of transgene, which happens more frequently with random integration of small transgenes,1,2,5 Targeted-CRISPR-LRS may be unable to account for all transgene breakpoints and/or boundaries. In this case, the broad sequencing approach of uHMW-gDNA WGS, paired with Targeted-CRISPR-LRS data, will better enable one to further define the complexity of transgene integration (Figs. 4 and 5), but with the understanding that most WGS data will not be used to resolve this locus. Lastly, to gain more confidence in mapping data, CRISPR-LRS defined transgene and genome breakpoints should be verified by PCR and Sanger Sequencing.
There are several benefits of mapping transgenes in animal models such as mice. First, mapping allows investigators to accelerate the generation of desired outcomes in a complex breeding scheme (e.g., floxed alleles with Cre-driver lines). Second, it enables quality design of genotyping assays for colony maintenance and zygosity determination. Lastly and arguably most important, it alerts one to confounding genetics caused by insertion of the transgene in a coding/noncoding gene locus or regulatory enhancer/element.1,2,5,10–12 Based on the data presented here, we envision CRISPR-LRS as the “go-to” method for mapping transgenes in the mouse genome and forecast its application in any transgenic organism with a reasonably annotated genome.
Footnotes
Acknowledgments
The authors wish to thank Akelia Wauchope-Odumbo and Carl Woodham at Oxford Nanopore Technologies (ONT) for their combined help with use of long-read nanopore sequencing.
Authors' Contributions
W.B.B. and J.M.M designed the study. A.Y., S.G., W.Z., and A.M.R. maintained mouse colonies. W.H. designed and prepared the Thy1-ECFP-VAMP2 transgene for the mouse line. W.B.B. performed the experiments. W.B.B. and J.M.M. analyzed and interpreted data. W.Z., X.L., and A.M.R. provided liver tissue. M.K.M and F.D. contributed to Figure concept/design and manuscript edits. W.B.B. and J.M.M. wrote the paper.
Availability of Data and Materials
Nanopore long-read sequencing data are available at NCBI Sequence Read Archive (SRA) under accession number PRJNA759232.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants HL138987 and HL147476 to Joseph M. Miano, NIH R01AG062655 to Ferenc Deak, and HL122686 and HL139794 to Xiaochun Long. Research in the laboratory of Weiping Han is supported by the Agency for Science, Technology and Research (A*STAR) Biomedical Research Council core fund, A*STAR Strategic Research Program (the Brain-Body Initiative, iGrants call ID #21718) and the A*STAR Use-Inspired Basic Research Award.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.