
Abstract
Target-AID, BE3, and ABE7.10 base editors fused to the catalytically modified Cas9 and xCas9(3.7) were tested for germline editing of the fruit fly Drosophila melanogaster. We developed a guide RNA-expressing construct, white-4gRNA, targeting splice sites in the white gene, an X-chromosome located gene. Using white-4gRNA flies and transgenic lines expressing Target-AID, BE3, and ABE7.10 base editors, we tested the efficiency of stable germline gene editing at three different temperatures. Classical Cas9 generating insertions/deletions by non-homologous end joining served as a reference. Our data indicate that gene editing is most efficient at 28°C, the highest temperature suitable for fruit flies. Finally, we created a new allele of the core circadian clock gene timeless using Target-AID. This base edited mutant allele timSS308-9FL had a disrupted circadian clock with a period of ∼29 h. The white-4gRNA expressing fly can be used to test new generations of base editors for future applications in Drosophila.
Introduction
CRISPR-Cas9 revolutionized genetic engineering and became a widely used tool in reverse genetics. Essentially, a complex of Cas9 protein and guide RNA (gRNA) is targeted to a specific DNA sequence where a double-strand break (DSB) occurs. Sequence recognition requires complementarity of 20–23 nucleotides between the gRNA and the target DNA, as well as a specific three-nucleotide protospacer adjacent motif (PAM), which is directly recognized by Cas9.1–4
After a DSB is created, the cleaved DNA is repaired by a nonhomologous end-joining (NHEJ) or microhomology-mediated end-joining (MMEJ) repair mechanism, which subsequently leads to gene disruption through insertions-deletions (indels), translocations, and other DNA rearrangements.5,6
While the generation of null mutants, often generated after the introduction of DSB with CRISPR-Cas9, is a powerful approach to uncover gene function, more delicate and controllable DNA modifications are usually required to decipher protein functions. Therefore, several approaches have been developed to make precise and specific changes in DNA sequence instead of the unpredictable DNA rearrangements resulting from NHEJ and MMEJ.
For example, along with the introduction of DNA breaks, a DNA template is provided for the homology-directed repair. Alternatively, prime editors rely on information provided by prime editing guide RNA. 7
A special group of genome-modifying enzymes are base editors, genetically engineered enzymes that can introduce point mutations without DSBs and without DNA template. Base editors consist of a DNA deaminase enzyme in combination with a catalytically impaired Cas9 nuclease that either generates only single-strand nicks (nCas9) or does not cleave DNA at all (dCas9). Further, some base editors contain additional enzyme domains, such as uracil glycosylase inhibitor (UGI), which help to maintain the mutation in the DNA.
Depending on the deaminase, two major groups of base editors are distinguished: cytosine base editors, which convert C/G pairs to T/A pairs, and adenine base editors (ABEs), which are responsible for the conversion of A/T to G/C pairs. The rat cytidine deaminase rAPOBEC1 was linked to dCas9 to produce a first-generation cytidine base editor (BE1), 8 which was further modified in several rounds to improve its efficiency. The third-generation cytidine base editor (BE3) had a mutation efficiency of nearly 75% and contained nickase-Cas9 (nCas9) and UGI as independent components. 8
Similarly, a cytidine deaminase from the sea lamprey PmCDA1 was harnessed with nCas9 in a system called Target-AID and achieved high efficiency of single nucleotide mutations.
9
In 2017, an ABE was produced by protein engineering of Escherichia coli adenine deaminase (TadA) that can convert A
However, genome targeting remained limited to the PAM, which is required for the specific Cas9. To overcome this challenge, Streptococcus pyogenes Cas9 (Sp Cas9) was engineered into xCas9 3.7 variant that has broader PAM compatibility with a binding sequence of NG, GAA, and GAT. 11
While the earlier-described base editors are conceptually established, the particular activity has been tested so far in some organisms, including animals such as zebrafish,12–15 mice,16–19 plants such as rice, wheat, and corn,20–23 and the insect silkworm Bombyx mori. 24
This study was designed as a test of base editors for stable germline transformation in Drosophila. Since we targeted a clearly visible marker, the white gene, we were able to compare the efficiency of three base editors and classical Cas9. Further, we tested the efficiency at three biologically meaningful temperatures. We found a relatively high level of mosaicism, confirming the previous reports of maternal deposition of the gRNA or Cas9 into fruit fly eggs.25–27 The fly line presented here, which expresses multiple gRNA constructs, may be used to assess the efficiency of new base editors in the future.
Finally, to prove the concept, we generated a new allele of timeless (tim), a well-established circadian clock gene, and characterized the new mutant. This mutant allele timSS308-9FL exhibited disruption of the circadian rhythm with a period of ∼29 h. In addition, a severe defect in temperature compensation was also observed when the timSS308-9FL allele was present as a hemizygote.
During our attempt to test base editing in Drosophila, two studies surfaced where in one study, the authors verified base editing in somatic cells using dBE2 cytidine deaminase 28 whereas in another study, the authors explored somatic and germline base editing by engineered versions of the cytidine deaminase dCBEevoCDA1 and dCBEevoAPOBEC1. 29
Material and Methods
Plasmids
To make Drosophila base editing constructs, we acquired four base editors from the Addgene plasmid repository: (1) xCas9(3.7)-BE3 construct that expresses the modified rat Apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 (rat APOBEC1); a cytidine deaminase (Addgene #108380) (hereafter referred as BE3) and (2) xCas9(3.7)-ABE(7.10) construct that expresses modified E. coli tRNA specific adenosine deaminase (E. coli tadA; Uniprot ID: P68398) (Addgene #108382) (hereafter referred as ABE7.10); both mammalian expression vectors originally made and later modified in David Liu's lab 11 with engineered Cas9 variant “x-Cas9(3.7)” broadening the PAM flexibility to GAA, GAT, and NG in addition to its canonical NGG. (3) pcDNA3.1_pCMV-nCas-PmCDA1-ugi PH1-gRNA(HPRT) construct, which expresses cytidine deaminase 1 from lamprey (PmCDA1) and gRNA (HPRT) in mammalian cells from Akihiko Kondo's lab 9 (Addgene #79620) (hereafter referred as Target-AID).
Base editor coding sequences were transferred to the pBFV-nosP-Cas9 construct from the Shu Kondo's lab 30 (Addgene #138402), which contains the nanos (nos) promoter and the “attB” sequence for PhiC31-based genome integration. See the Supplementary Material and Methods for details of cloning and Supplementary Figures S1 and S2 for schematic maps of plasmids.
Two multiple gRNA constructs derived from pCFD5 multiplex gRNA plasmid (Addgene #73914) containing the Drosophila U6.3 promoter, the “attB” sequence, and vermilion as phenotypic marker were used in this study. To construct the white-4gRNA plasmid, four gRNA sequences (Table 1) with CRISPR base editing window at the exon-intron boundaries were integrated into 43–73 bp primers flanking pCFD5 plasmid (see Table 2 for primers and Supplementary Fig. S3 for plasmid map).
List of guide RNA sequences

Primers for Gibson cloning
A total of three fragments were prepared by PCR using the earlier-mentioned primers and the BbsI-digested pCFD5 plasmid as template. These three fragments were cloned into the BbsI-digested pCFD5 vector using the Gibson cloning kit (New England Biolabs) (for detailed protocol see Ref. 31 ).
For targeting the tim gene, three gRNA sequences (Table 1) with CRISPR base editing window in exon 4 of the tim gene were cloned into the BbsI-digested pCFD5 using identical strategy (PCR amplification, Gibson assembly) as for the white-4gRNA plasmid. See Table 2 for the primers and Supplementary Figure S4 for the map of tim-3gRNA plasmid.
Drosophila genetics
Plasmids were injected into y[1] M{vas-int.Dm}ZH-2A w[*]; P{y[+t7.7] = CaryP}attP2 carrying docking site on the third chromosome (WellGenetics, Inc., Taiwan) and founder flies were balanced with TM6C, after which homozygous lines were established. For white gene editing, the crossing scheme depicted in Figure 1 was used. First, 10 single crosses were carried out for each base editor, in which virgin females homozygous for the base editor were crossed with males homozygous for the white-4gRNA construct.

Schematic description of here-tested constructs and a crossing scheme illustrating the targeting of the white gene in flies.
From the progeny, ∼20 virgin flies (2 from each vial) that were heterozygous for the base editor and white-4gRNA were collected and individually crossed with Canton-S males. In the next generation, males with mutant eyes were collected, snap-frozen, and stored at −80°C for later molecular verification. The same protocol was repeated in four biological replicates for each base editor at 18°C, 25°C, and 28°C.
In parallel, a comparable crossing scheme was performed with nos-Cas9, where virgin flies used were y2cho 2 v 1 ; P{nos-Cas9, y+, v+}3A/TM6C, Sb Tb. To test whether the low editing efficiency of base editors and nos-Cas9 might be caused by the nanos promoter, we performed an experiment utilizing vas-Cas9 transgenic line marked with 3xP3-GFP (Bloomington Drosophila Stock Center #51324, w[1118]; PBac{y[+mDint2] = vas-Cas9}VK00027).
The crossing scheme remained the same as mentioned earlier, except that 20 virgin flies heterozygous for the vas-Cas9 and white-4gRNA were collected (instead of 40) and individually crossed together.
For tim gene editing, three single crosses were set up at 25°C by using homozygous males with tim-3gRNA and homozygous virgin females with nos-Target-AID (both transgenes are located on chromosome III). Heterozygous nos-Target-AID/tim-3gRNA female progeny were crossed to If/CyO males.
From their progeny, males and virgin females with CyO balancer were collected (tim is located on the second chromosome, and individually crossed with If/CyO (∼15 single crosses)). From the progeny of each individual cross, +/CyO males and virgins were collected to establish unique lines, each with identical second chromosomes balanced by CyO. The mutation was identified by sequencing the target region of tim in homozygous individuals.
Sequencing
For the white gene editing, we randomly selected ∼15 single white-eyed males (5 from each temperature, if possible) from the individual cross for each base editor and canonical Cas9. DNA was isolated using the squish protocol, white gene regions PCR-amplified and sequenced as described earlier. 32 For tim gene editing, 24 males (each representing a different line) homozygous for the second chromosome were individually squished and the targeted region of tim was PCR-amplified, purified, and sequenced (primers in Supplementary Table S1).
Splice site prediction
Splice Site Prediction by Neural Network of Berkeley Drosophila Genome Project was used to predict splice sites in the white gene of selected mutants. The threshold score for 5′ and 3′ splice site prediction was set to 0.1 to predict any splice site even with the lowest score. However, any prediction score above 0.4 is considered optimal.
Genotyping mosaic males
To determine the genotype of males with mosaic eyes, individual flies were homogenized in 50 μL of squishing buffer. Transgene-specific primers were used in PCR, products electrophoretically separated, and the presence/absence of bands indicated whether the white-4gRNA construct or base editor/Cas9 construct was present. Control PCR was run with the original transgenic lines serving as a template to confirm the specificity of all primers.
Drosophila activity monitoring and analysis
CO2-anesthetized, 3-day old males were housed in 5 mm glass tubes containing food (5% sucrose, 2% agar) and tubes were loaded into the Drosophila Activity Monitor system 2 (DAM2; Trikinetics, Waltham, USA). Flies were entrained for 5 days in a light:dark cycle of 12 h (LD, 12:12), after which the lights were turned off and constant darkness (DD) was maintained for 12–15 days.
The activity was measured at 17°C, 20°C, 25°C, and 28°C. The first 10 days of DD were considered when calculating the free-running period (tau, τ) and the last 2–5 days served as a reference for the health/survival of the fly. From the activity data, double-plotted actograms were made using the ActogramJ plugin 33 and the Lomb-Scargle periodogram analysis was performed to find out the τ of the flies. Individual τ values and percentage rhythmicity data were plotted (GraphPad; Prism).
Results
Targeting the white gene
To test the efficiency of editing, disruption of the Drosophila white gene was used. This X chromosome-located gene facilitates efficient detection of mutation in males. Balancer chromosomes in Drosophila allow a reliable combination of gRNA and base editor transgenes, resulting in male mutant progeny after two genetic crosses (Fig. 1). Here, we explored cytidine (BE3, Target-AID) and adenine (ABE7.10) base editing in Drosophila, by driving their expression from germline-specific nanos promoter.
All three base editor constructs contain a nCas9, and BE3 and ABE7.10 constructs also contain engineered xCas9(3.7) variant permitting recognition to different PAM motifs such as NG, GAA and GAT 11 (Fig. 1A; Supplementary Fig. S2).
However, creating a null mutation by introducing a premature stop codon (TGA, TAA, or TAG) is possible only with cytidine base editors, whereas de novo production of any stop codon is not achievable with ABEs (stop codon can be obtained only from another stop codon). Therefore, to be able to use identical gRNAs for all tested base editors, we targeted the 5′ and 3′ splice recognition sites in the white gene with the aim of preventing the proper splicing of transcribed RNA to mRNA. To ensure a severe impact on the resulting protein, the exon boundaries of the first three exons were targeted (Fig. 3A).
The clearly visible eye color allowed us to phenotypically analyze around 20,000 male flies for each tested base editor, where nos-Cas9 served as a positive control. Further, we assessed the editing capacity at three temperatures. Flies with altered phenotype fell into two categories: complete eye color changed to white (sometimes slightly yellow), and flies with a mosaic distribution of white and red (wild-type) color. For evaluating the efficiency, we plotted the frequency of mutant flies identified per total number of flies (Fig. 2A, B, Supplementary Table S4) and the percentage of vials (that is offspring of a single cross) with at least one mutant fly (Fig. 2C, D, Supplementary Table S5 and Supplementary Fig. S5).
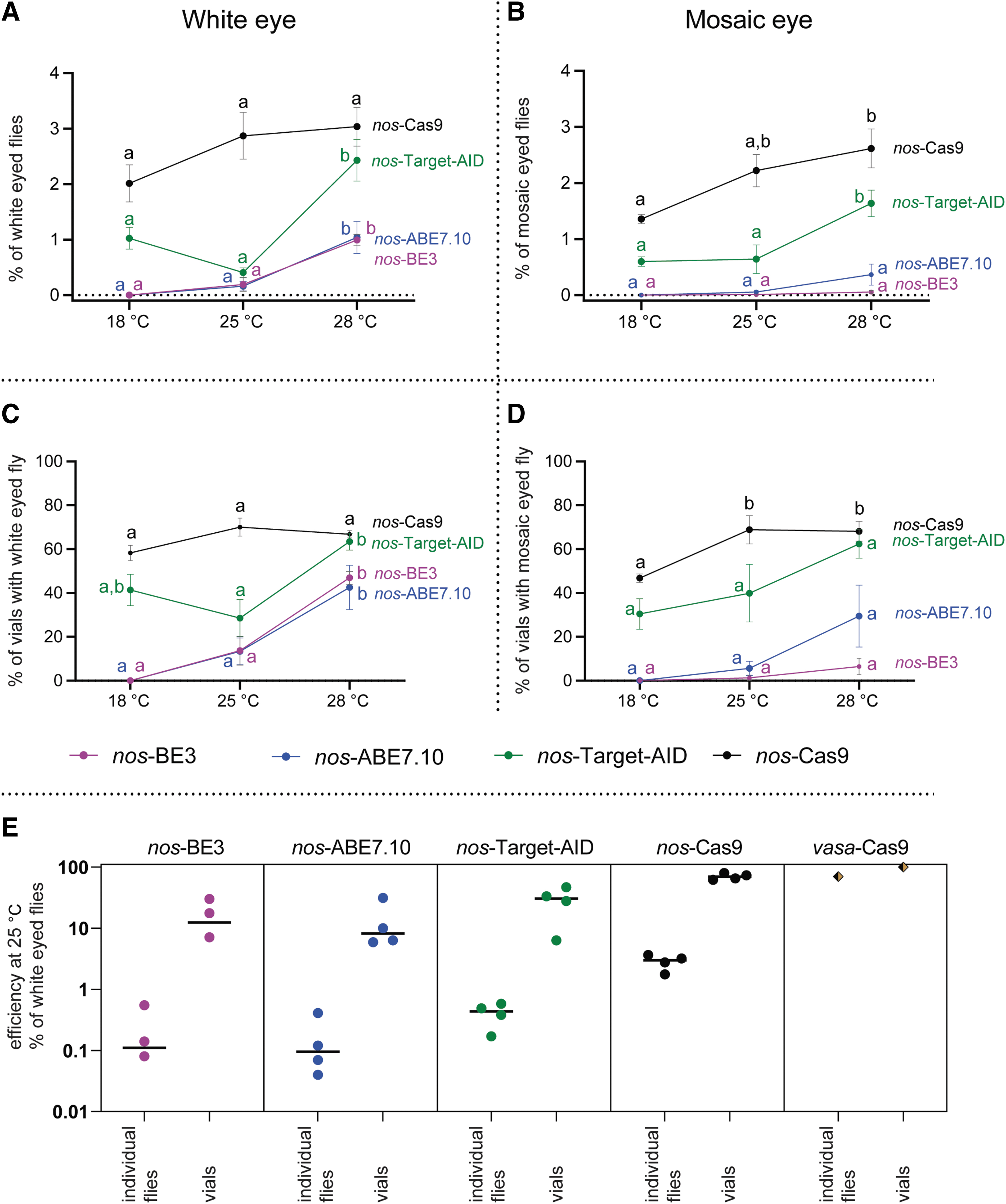
Efficiency of white gene mutagenesis at three temperatures using the Drosophila germline expressed (nanos promoter) base editor and the canonical CRISPR Cas9 editor.
Comparison of the efficiencies with vasa-Cas9
To test whether the editing efficiency is influenced by the white-4gRNA construct, additional cross was performed with a highly active vasa-Cas9 line. As illustrated in Figure 2E and Supplementary Table S6, the data exclude the possibility that the low efficiency observed with the base editors and nos-Cas9 are due to inefficient gRNAs.
The efficiency is affected by the temperature
At an ambient temperature of 25°C, Target-AID was the most efficient base editor in generating white-eyed flies (0.4% of flies; 29% of vials/crosses), whereas the efficiency of BE3 and ABE7.10 was comparable (0.16–0.19% of flies; 13% of crosses). Reference nos-Cas9 generated approximately seven times more mutants than Target-AID (2.87% of flies; 70% of crosses; Fig. 2A, C; Supplementary Tables S2 and S3).
Because Drosophila does not control its body temperature, we compared the efficiency of base editors at 18°C, 25°C, and 28°C. At all temperatures, the trend of efficiency was similar (nos-Cas9 > Target-AID > BE3/ABE7.10). However, comparing the efficiency of the same editor at three temperatures showed different trends. nos-Cas9 was comparably efficient at all three temperatures (the differences were not statistically significant).
The efficiency of BE3 and ABE7.10 increased with temperature, whereas no editing was detected at 18°C and efficiency increased approximately fivefold from 25°C to 28°C. Target-AID was most efficient at 28°C, whereas efficiency was lowest at 25°C and intermediate at 18°C (Fig. 2A, C; Supplementary Tables S2 and S3).
Mosaicism
We identified a substantial number of males in which the compound eyes exhibited a distinctly mosaic distribution of pigment (Figs. 2B, D and 4B–E). In general, the trends were like those observed for white-eyed fly abundance. The highest frequency was observed for nos-Cas9 and then for Target-AID. While white-eyed flies were produced with comparable efficiency by BE3 and ABE7.10, mosaics were produced more frequently with ABE7.10 than with BE3. The frequency increased with temperature for each base editor and even for Target-AID (Fig. 2B, D).
Molecular changes introduced by base editors and nos-Cas9
White gene target regions were sequenced from ∼15 males for each base editor and nos-Cas9. When possible, five males were selected for each temperature. Because the regions with four target sites had to be amplified in three separate PCR reactions, we assembled the outcomes into one merged sequence that allows to identify and interpret co-occurrence of modifications (Supplementary Figs. S6–S9). A summary of the sequencing is depicted in Figure 3B.
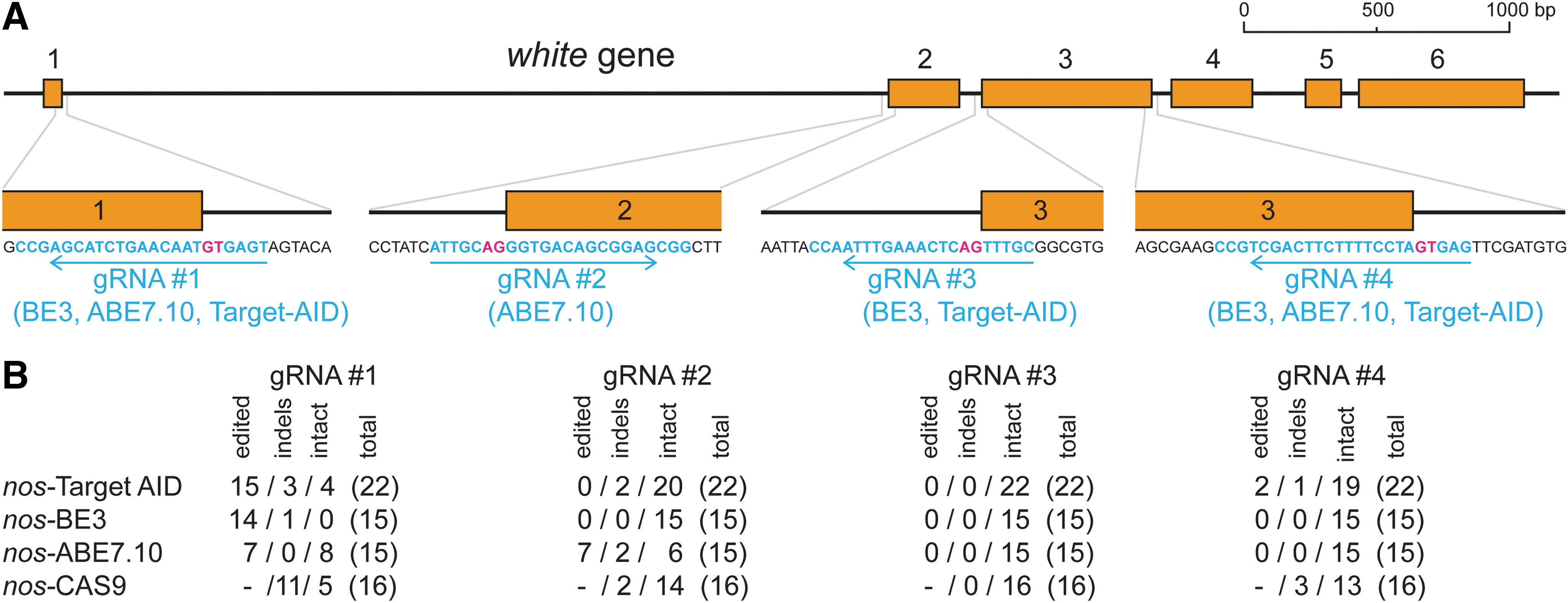
Details of the white gene editing.
The efficiency varied between target sites: The region targeted by gRNA1 was edited most frequently, whereas no editing was detected for region targeted by gRNA3. Further, a few deletions were also observed.
Sequence analysis revealed that the 5′ splice donor site was precisely modified by Target-AID at #1 and #4 (Supplementary Fig. S6) whereas BE3 modified it only at #1 (Supplementary Fig. S7; note that the reverse DNA strand was targeted in #1 and #4, therefore C-to-T modifications are depicted as G-to-A in the Supplementary Figs. S6 and S7). Region #1 illustrates the difference in editing specificity: within a five-nucleotide window, Target-AID modified three cytidines (six flies), two cytidines (three flies), or only one cytidine (four flies).
In one fly, a cytidine located seven nucleotides upstream of the window was also modified. BE3 modified either two cytidines (13 flies) or one cytidine (1 fly). Moreover, two C-to-G changes in the editing window were also modified by Target-AID (Supplementary Fig. S6).
ABE7.10 precisely modified “A” to “G” in the 5′ splice donor site at #1 (reverse strand was targeted) and in the 3′ splice acceptor site at #2 (Supplementary Fig. S8). Contrary to cytidine base editors, in the case of ABE7.10, additional “A” at #1 was not modified despite it falling in the editing window (Supplementary Fig. S8).
Canonical CRISPR-Cas9 editing destroyed the target region due to several indels in #1, #2, and #4 (Supplementary Fig. S9; Fig. 3B). Contrarily, the Target-AID, BE3, and ABE7.10 scarcely disrupted the target DNA in regions #1, #2, and #4, resulting in very few indels (Supplementary Figs. S6–S8; Fig. 3B). Notably, the indels generated by the base editors differed from those of the nos-Cas9, comprising mainly deletions, whereas the Cas9-created indels carried insertions and deletions (Supplementary Figs. S6–S9).
A small subset of the flies with completely changed eye color was slightly yellowish. We sequenced seven such flies generated with Target-AID and one with nos-Cas9 (labelled “yellow” in Supplementary Figs. S6 and S9). Three of them contained short deletion, whereas five of them (all from Target-AID) contained only one nucleotide substitution downstream of the splice site resulting in a prediction of less efficient splice recognition site by Splice Site Prediction Tool Neural Network (0.27 or 0.66 instead of the 0.88 in the original sequence; Supplementary Fig. S10).
A subset of F2 males exhibited a distinct mosaic eye. When the white gene was PCR amplified from selected mosaic males and sequenced, we could see clean chromatograms that abruptly changed to a chromatogram in which multiple peaks overlapped (Supplementary Fig. S11). The position where this change occurs corresponds exactly to the predicted editing site. Since some flies contained deletions even after base editors were applied, we assume that a subset of the white genes contains a deletion that leads to the observed sequencing pattern.
Genotype of mosaic males
Males of F2 generation (Fig. 1E) should contain either a gRNA-encoding construct or a construct with base editor, but never both (all constructs landed at the identical docking site on the third chromosome, so that even recombination in the females [Fig. 1D], could not lead to the presence of both constructs in F2 males).
To clarify the genotype of the F2 males, PCR was used to detect the presence of gRNA and base editor constructs. First, we indeed confirmed the presence of just one construct in each F2 male. In all three base editor and in nos-Cas9 crosses, either the gRNA construct or the base editor construct (Cas9) was detected. Enough flies with mosaic eyes were available only for Target-AID and nos-Cas9; their analysis indicates approximately equal presence of gRNA or base editor (Cas9) constructs (Fig. 4G). Accordingly, either the gRNA or the base editor protein (or its mRNA) must be deposited maternally into the egg.
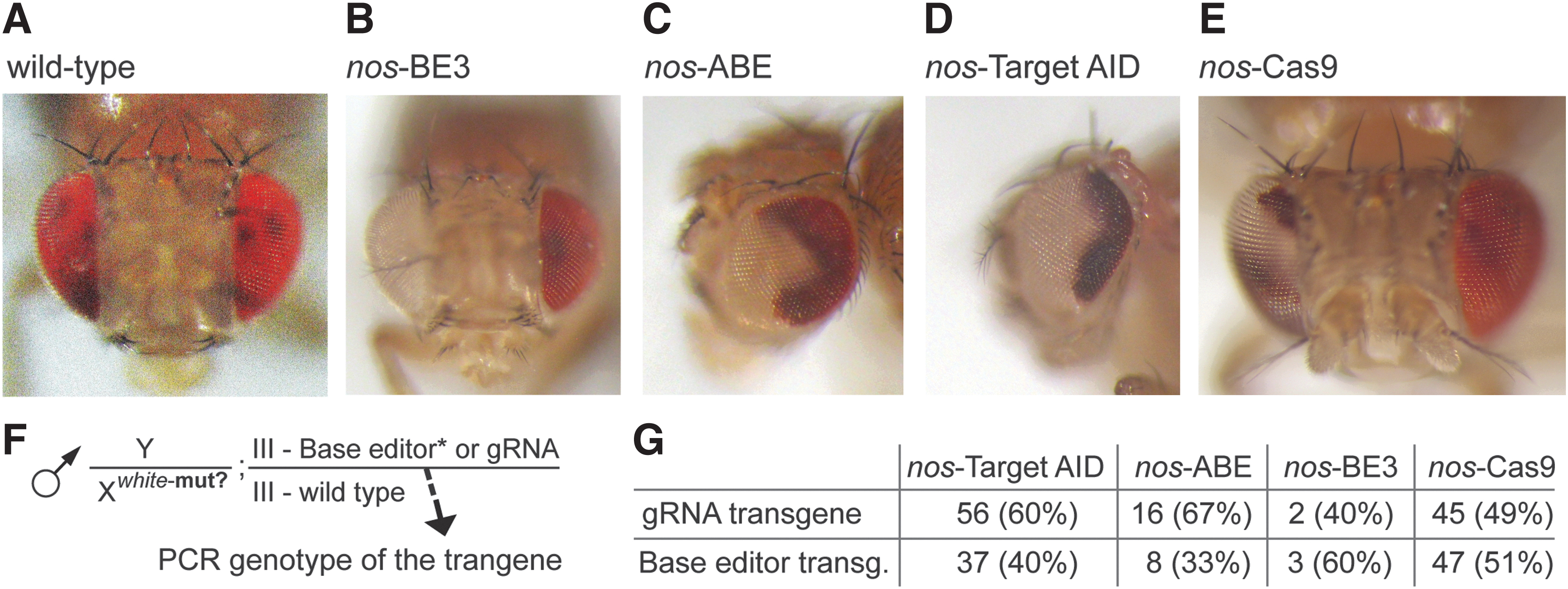
Eye color mosaicism was detected in a subset of F2 males.
Targeting circadian clock gene tim
Given the successful modification of a gene with a clearly visible phenotypic change, we tested the applicability of the gene editing protocol for the introduction of mutations that might be informative for a specific research question. The target was tim, a gene that is a well-established component of the circadian clock,34–36 but because of its length, several regions have not previously been targeted experimentally.
First, we performed in silico analysis of the Drosophila TIM protein across various insect species and identified a highly conserved region similar even in the most basal insect Thermobia domestica (Fig. 5A). Moreover, a few point mutants with divergent circadian phenotypes have been described in this region, but the regulatory mechanism is not completely understood32,37,38 (Fig. 5A).
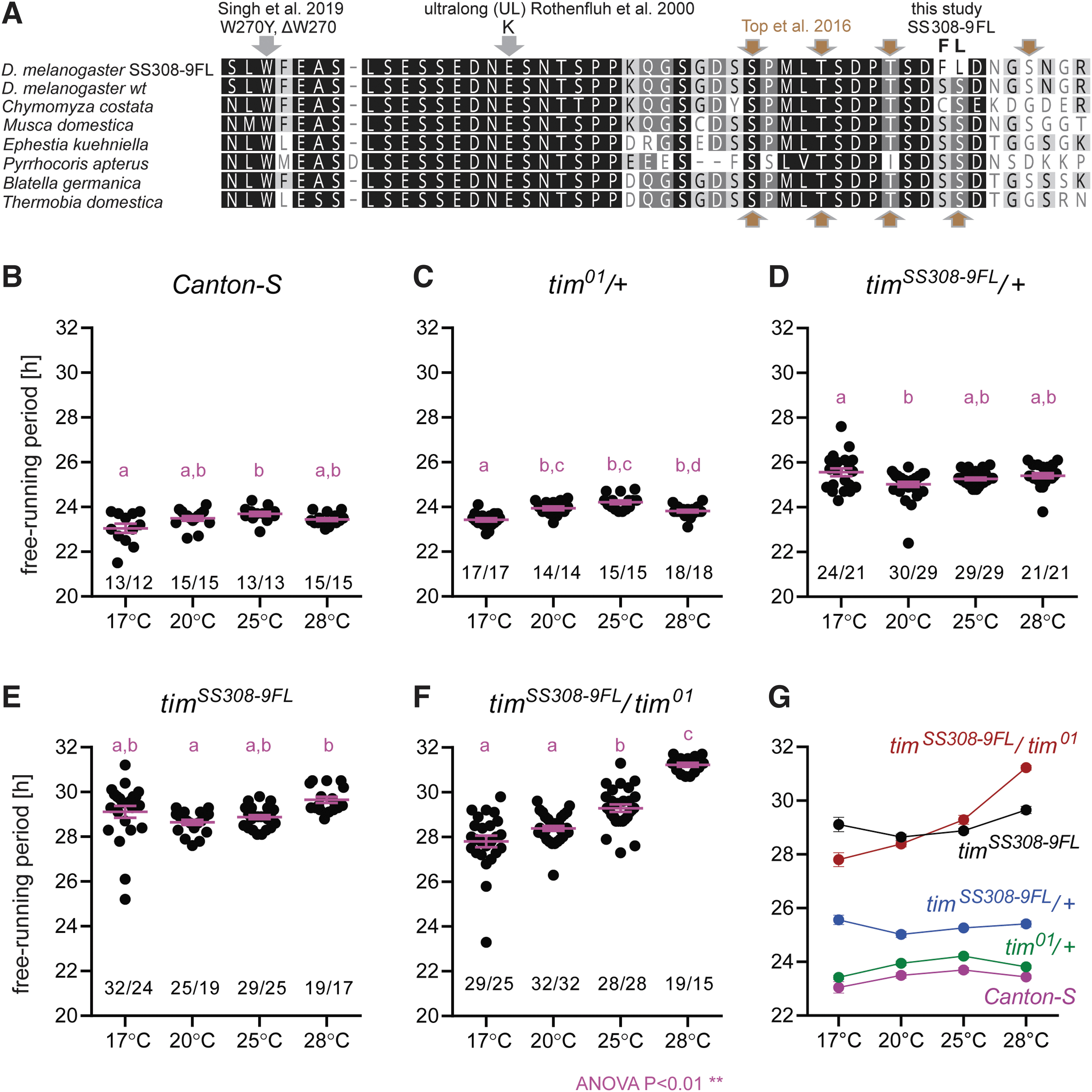
Functional analysis of the Target-AID-edited timeless mutant.
Therefore, we targeted two highly conserved serines (S) at positions 308–309 in TIM (numbered according to the L-TIM isoform; 1421 aa) with the DNA coding sequence of TCC and TCG (Fig. 5A; Supplementary Fig. S12) using the Target-AID. Finally, from the mutagenic screen, we sequenced 24 lines and identified a base-edited tim mutant (∼4% editing efficiency) with the modified DNA coding sequence of TTT and TTG encoding for phenylalanine (F) and leucine (L) (timSS308-9FL) (Fig. 5A top; Supplementary Fig. S12).
To determine the impact of the mutation on the Drosophila circadian clock, we analyzed the locomotor activity of the timSS308-9FL mutant at four different temperatures (17°C, 20°C, 25°C, and 28°C). As shown in Figure 5B, the free-running period (tau, τ) of wild-type flies (canton-s) is nearly 24 h (τ ∽ 23.5 h) over the entire physiological temperature range of Drosophila.
In agreement with previous studies,32,39 even the single copy of wild-type tim is sufficient to keep the clock ticking at the same pace at 17°C, 20°C, 25°C, and 28°C (tim01/+; τ ∼ 23.5 h) (Fig. 5C, G; Supplementary Fig. S13). Surprisingly, the base-edited timeless mutant timSS308–9FL allele was found to be dominant over wild-type tim and slowed down the circadian clock by nearly 1.5 h (timSS308-9FL/+ heterozygote; τ ∽ 25 h; Fig. 5D, G; Supplementary Fig. S13).
In timSS308–9FL homozygotes, the circadian clock slowed by as much as 5.5 h (τ ∽ 29 h) compared with wild-type flies (Fig. 5E; Supplementary Fig. S13). Further, timSS308-9FL hemizygotes showed impaired temperature compensation, with the clock running faster at 17°C (τ ∽ 28 h) and slower at 28°C (τ ∽ 31 h) (Fig. 5F, G; Supplementary Fig. S13).
Discussion
The fact that base editors do not generate DSB makes them a promising tool for gene therapy. Originally, base editors were developed in mammalian cell culture assays with the intention of rectifying single nucleotide errors in human genetic diseases. Nonetheless, given their potential as a scarless gene editing tool, CRISPR base editors were adapted in several model organisms such as mice,16–19 zebrafish,12–15 the silkworm B. mori, 24 and various plant species.20–23
However, the base editors remained to be tested in Drosophila melanogaster, a key model indispensable for insect physiology and human translational research.40,41 Therefore, we introduced two cytidine (Target-AID, BE3) and one adenine (ABE7.10) base editors in the Drosophila system.
During our endeavor, one study addressed dBE2 (second-generation cytidine base editor) in Drosophila somatic cells, 28 whereas another study explored somatic and germline base editing of the cytidine deaminases dCBEevoCDA1 and dCBEevoAPOBEC1. 29
Notably, even the highest efficiencies of our base editors were very low (0.9–2.4% at 28°C; Supplementary Table S2) contrasting with 70–95% of dCBEevoCDA1 and dCBEevoAPOBEC1. 29
Since epigenetic features or local molecular environment affects targeting by Cas9 regardless of prediction score,42–44 we tested the efficiency of white-4gRNA to rule out that the construct produces inefficient gRNAs (Fig. 2E). The editing efficiency is further impacted by expression of Cas9/base editor, that is, the combination of a particular promoter in the construct and the landing site.45,46 Indeed, some nos-Cas9 lines (i.e., CAS0003) have low activity whereas the same construct inserted elsewhere in the genome is more efficient. 46
Since here used attP2 landing site supports good transgene expression, the low efficiency might be connected to the base editor design. Possible explanations include Cas9 nuclease modifications to either curb indels or broaden the PAM recognition sequence. Although xCas9(3.7) nickase-based editors are less effective than the canonical Cas9-derived target-AID, additional changes in these base editors outside of Cas9 could contribute, such as N- or C-terminal orientation of the deaminase affecting the editing window, spacers affecting the structural flexibility of the deaminases, and the dosage of nuclear localization signal sequence.
Depending on the intent of the experiment (efficiency vs. lethality), CRISPR base-editors expressed from different drivers are required. Target-AID, BE3, and ABE7.10 expressed from nanos promoter serve the purpose of germline restricted base editing.
In our study, we compared the efficiency of base editors expressed in flies at three temperatures (18°C, 25°C, and 28°C) from the nanos promoter (Supplementary Fig. S3) and the tRNAs flanking multiple gRNAs (targeting distinct exon-intron regions of the white gene) from the RNA Pol III promoter U6 (Supplementary Fig. S1). The CRISPR base editors were functional at 25°C (Fig. 2A; Supplementary Fig. S4A).
However, more male flies with white eyes were found at 28°C by CRISPR base editor mutagenesis, where editing by Target-AID was nearly threefold more efficient than BE3 and ABE7.10 (Fig. 2A; Supplementary Fig. S4A). Similarly, with increasing temperature, Doll et al. also observed higher C to T editing efficiency, where editing by dCBEevoCDA1 was nearly two folds more efficient than dCBEevoAPOBEC1 at 28°C. 29
A similar temperature trend was also described with canonical CRISPR-Cas9 mutagenesis where the mutation efficiency increased several fold when Arabidopsis plants were subjected to heat treatment at 37°C.47,48 Moreover, the effect of hyperthermia on CRISPR Cas9-mediated genome editing was observed in mammalian cell lines with most DNA modifications at 39°C. 49 Interestingly, we did not find a similar trend in our canonical CRISPR Cas9 mutagenesis in Drosophila (Fig. 2A; Supplementary Fig. S4A).
Further, an increase in knock-in and knock-out efficiency of the CRISPR-Cas9 tool was observed when the African clawed frog Xenopus laevis embryos were incubated for a specified time at a lower temperature after microinjection. 50 However, at 18°C, the efficiency of canonical CRISPR-Cas9 decreased in Drosophila (Fig. 2A; Supplementary Fig. S4A).
Strangely, the efficiency of Target-AID increased at 18°C compared with 25°C, making it the candidate base editor that can be used at low temperatures (Fig. 2A; Supplementary Fig. S4A). dCBEevoCDA1 (engineered version of the lamprey cytidine deaminase) used in the concurrent study on Drosophila was also functional at 18°C, however its efficiency decreased compared with 24°C. 29 Perhaps, the functioning of the Target-AID at a lower temperature can be attributed to the higher activity of lamprey cytidine deaminase at a lower temperature (the host organism, sea lamprey Petromyzon marinus, lives in cold water). 51
Notably, we obtained a few deletion mutants from Target-AID, BE3, and ABE7.10 (Fig. 3B; Supplementary Figs. S6–S8). Likewise, a small number of indels were also reported in the original studies in which these base editors were developed (typically ≤1% for BE3, <0.3% for Target-AID, and ≤0.1% for ABE7.10),8–10 in the silkworm B. mori (≤0.6% for BE3) 24 and in the study describing base editing in Drosophila (≤0.5% for dCBEevoAPOBEC1 and ≤2.3% for dCBEevoCDA1). 29
Such indels result from rare circumstances when the base excision repair pathway gets activated first, leading to the removal of the deaminated base and creation of a nick. Consequently, this causes DSB that leads to insertion and deletion-prone NHEJ.52,53 Over the coming years, cytidine base editors have been improved to decrease the indels.54–56 Thus, possible base editors with more precise editing capacity are available. The versions of base editors selected here combine some level of mutagenic capacity with precise editing so that they can be used to generate a wide range of mutants in conserved and less explored genomic regions.
While the original purpose of base editors requires editing to be as precise and focused as possible, base editors also open the possibility of becoming a tool for targeted mutagenesis. In this case, a restricted but broader window of up to tens or even hundreds of nucleotides would provide remarkable benefits. Although some base editors of this type have already been developed, 57 this line of research is still relatively underexplored. Another strategy that leads to a broader editing window is the simultaneous use of multiple gRNA.
Originally, multiple gRNA constructs in combination with Cas9 provided highly efficient gene deletions, even in a tissue-specific manner. 31 Combining base editors with multiple gRNA, several different targets can be edited with a possibility of different combinations of edited variants. In addition, editing diversity can be increased by using different base editors with the same set of gRNAs.
As we show here, the same gRNA leads to distinct editing patterns depending on the base editor; in the case of Target-AID, even various degrees of editing were observed at the same locus. Notably, simultaneous use of two gRNA spaced ∼50 bp apart broadened the region modified by cytidine base editors. 28
Like the F2 generation mosaic-eyed flies encountered in this study (Figs. 1E and 4F), various studies have reported non-mendelian trespassing of CRISPR-Cas9 components during oocyte maturation25–27 which should be considered when doing gene manipulations using CRISPR-Cas9 and base editors.
As an example of a successful application of base editing, we created a new mutant of the tim gene. In principle, we used a similar approach as before, 32 except that Cas9 was replaced by Target-AID. The obtained mutation extended the free-running period similarly to the 3A mutant combining three substitutions T305A/S309A/S313A that prevent phosphorylation. 38
The 3A mutant had 27-h-long behavioral rhythms, whereas in timSS308–9FL homozygotes, the circadian clock was slowed to ∽29 h at 25°C. Interestingly, the obtained mutant showed a dose-dependent defect in temperature compensation. A similar trend was observed for several other (but not all) tim mutants.32,58 However, temperature compensation is not limited to the tim gene.
Temperature-dependent changes in the free-running period have been observed in the period gene59–61 and in K224D mutants of doubletime, 62 a casein kinase I homolog responsible for PERIOD phosphorylation. Therefore, mutagenesis of clock proteins should be performed with the goal of identifying novel alleles responsible for temperature compensation. Drosophila tim gene studied here appears to function differently in some non-model insects as revealed from loss of function mutants.63,64 Thus, precise editing tools that go beyond loss-of-function are needed in these species to decipher underlying differences. Base editors might be a suitable tool for such tasks.
Conclusions
Base editing expands the already broad toolkit of reverse genetics in Drosophila. In addition to being able to make precise gene changes in narrow windows, base editors can also serve as mutagenic tools, in which case a wider window or simultaneous expression of multiple gRNAs is preferable. The three base editors tested here demonstrate that distinct modifications can be achieved with identical gRNAs. The white multiple-gRNA fly (white-4gRNA) can serve as a reporter line to measure the efficiency of new base editors in the future. Finally, the efficiency of base editing in Drosophila is temperature dependent.
Footnotes
Acknowledgments
The authors thank Vlastimil Smykal for photography assistance, Shu Kondo for nos-Cas9 expressing flies and pBFV-nosP-Cas9 (Addgene #138402), Tomas Dolezal for vas-Cas9 expressing flies (BDSC #51324), Port and Bullock for pCFD5 multiplex gRNA plasmid (Addgene #73914), David Liu and Akihiko Kondo for plasmids encoding base-editors (Addgene #108380, 108382, and 79620), and two anonymous reviewers for constructive comments.
Authors' Contributions
Conceptualization of the study was done by D.D.; formal analysis was performed by N.T., A.H., and V.B.; investigation was done by N.T., A.H., and V.B.; original draft of the article was written by N.T.; writing, review, and editing of the article was done by N.T and D.D.; funding acquisition was done by D.D.; supervision of the study was done by D.D. All authors approved the final article.
Disclaimer
The article has been submitted solely to this journal and is not published, in press, or submitted elsewhere.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was entirely supported by the Czech Science Foundation (GACR) Grant Number 22-10088S to D.D.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.