
Abstract
Genome-edited human-induced pluripotent stem cells (iPSCs) have broad applications in disease modeling, drug discovery, and regenerative medicine. Despite the development of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system, the gene editing process is inefficient and can take several weeks to months to generate edited iPSC clones. We developed a strategy to improve the efficiency of the iPSC gene editing process via application of a small-molecule, trichostatin A (TSA), a Class I and II histone deacetylase inhibitor. We observed that TSA decreased global chromatin condensation and further resulted in increased gene-editing efficiency of iPSCs by twofold to fourfold while concurrently ensuring no increased off-target effects. The edited iPSCs could be clonally expanded while maintaining genomic integrity and pluripotency. The rapid generation of therapeutically relevant gene-edited iPSCs could be enabled by these findings.
Introduction
Combining genome editing with human-induced pluripotent stem cells (iPSCs) constitutes a major advance in modern biotechnology. The pairing of gene editing technologies with human iPSCs for disease modeling overcomes the problem of animal models and human immortalized cell line models, which do not accurately represent the genetic background or cellular physiology of the patient.1–5 Human iPSC-based models are thus a valuable resource for studying disease mechanisms,6–10 screening potential new therapeutics, and testing toxic side effects of drug treatments.11–15 Moreover, performing gene editing on patient-derived iPSCs before differentiation enables the generation of isogenic iPSC lines, which can function as a precise control for studying the genetic disease model of interest.16–18 In addition, edited iPSCs can be used for cellular therapies19–24 and could overcome the problem of immune rejection.
However, the process of gene editing iPSCs is typically laborious and inefficient involving multiple steps, requiring lengthy cell culture periods, drug selection, and several clonal events (i.e., gene targeting, and subsequent genetic excision of a selection cassette25–29 ). The selection of rare targeted clones in iPSCs can be particularly difficult because they grow poorly as single cells.30,31 Several recent approaches have been developed to improve the specificity, efficiency, and versatility of the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 gene editing process by optimizing the structure of guide RNAs (gRNAs),32–34 or by using modified nuclease strategies,35–39 natural and engineered Cas9 variants,40–43 and small molecules.44–46 However, these techniques still generate heterogeneous human cell populations that require time-consuming and laborious steps, significant subsequent characterization steps for on-target edits, off-target effects, genomic integrity, and unknown disease-causing mutations or risk variants.39,47
Therefore, there is a need to overcome these limitations for enabling the use of gene-edited iPSCs for cell therapies.
Recent work indicates that chromatin structure of the target can have significant effects on Cas9 binding and gene editing efficiencies,7,48–52 thus leading to variation in targeting efficiency and choice of DNA repair pathway53–57 even between the same target sequences in varied chromatin contexts in different cell types as well as in vitro and in vivo. Studies have shown that closed chromatin can negatively affect Cas9 binding58–62 or delay CRISPR-Cas9 mutagenesis, 63 nucleosomes can block or present a hurdle for Cas9 access to DNA,64–67 and active transcription in open chromatin state can directly stimulate DNA cleavage by influencing Cas9 release states in a strand-specific manner.50,68 Moreover, off-target binding of Cas9 to target seed sequences has been shown to correlate with DNase I hypersensitivity sequences and inversely correlates with CpG methylation sites.61,63,69
Although these studies have shown that the chromatin structure can play a key role in gene editing and strategies have emerged to manipulate the chromatin state to modulate gene editing efficiency,58,70–74 they are primarily based on immortalized cell lines or cancer cell lines, which may not be clinically relevant. Moreover, although the chromatin structure has been shown to impact off-target effects,61,63,69,75 the impact of chromatin structure on off-target effects in the context of gene editing has not been well characterized.76,77
In this study, we address these challenges using a two-part strategy to study the impact of chromatin structure on gene editing outcomes in iPSCs. For our proof-of-concept study, we use a small molecule known to promote open chromatin state, called as trichostatin A (TSA; Class I and II histone deacetylase inhibitor or HDAC inhibitor). 78 We used TSA as it is one of the few FDA-approved HDAC inhibitors. Moreover, the TSA-induced changes in chromatin state can be easily assessed visually using nuclear imaging.78–80 First, we used live, in situ nuclear imaging to quantify the TSA-induced change in global chromatin state of iPSC nuclei, termed as chromatin condensation.79,81–83
Second, we performed extensive characterization to assess the impact of TSA on gene editing outcomes, that is, on-target efficiencies, off-target effects, pluripotency, and genomic integrity of the iPSCs. We observed that appropriate TSA treatment of iPSCs decreased chromatin condensation, increased the gene editing efficiency by twofold to fourfold while simultaneously ensuring genomic integrity and without any detectable increased off-target effects. Overall, we developed a strategy for rapid and in situ imaging-based identification of iPSCs amenable to gene editing and modified the gene editing process to generate edited iPSCs in a precise and efficient manner.
Materials and Methods
Cell culture
Mono-allelic monomeric Enhanced Green Fluorescent Protein (mEGFP)-tagged HIST1H2BJ human iPSCs (AICS-0061-036) were obtained from Allen Institute for Cell Science. This cell line was derived from the WTC parental line (GM25256) released by the Conklin Laboratory at the J. David Gladstone Institute. iPSCs were maintained in mTeSR1 medium on Matrigel ® (WiCell)-coated tissue culture polystyrene plates (BD Falcon). Cells were passaged every 4–5 days at a ratio of 1:8 using ReLeSR™ solution (STEMCELL Technologies). All cells were maintained at 37°C in 5% CO2 and tested monthly for possible mycoplasma contamination.
Chemical reagents
TSA (Millipore Sigma) was resuspended in dimethyl sulfoxide at a stock concentration of 2 mg/mL, respectively. Aliquots were then stored in −20°C. The purity of the inhibitors was assessed by Millipore Sigma (>99%).
Cell viability
Flow cytometry was performed using Ghost Dye™ Red 780 viability dye (Tonbo Biosciences) to determine the dose range of the TSA that can be administered to the cells without affecting cell viability. iPSCs were seeded at a density of 15,000 cells per well, cultured overnight, and incubated with TSA in 96-well plates for 24 h. The next day, cells were singularized using Accutase™ (STEMCELL Technologies), washed with phosphate-buffered saline (PBS), centrifuged at 300 g for 5 min, and Ghost Dye Red 780 viability dye was added at 1:1000 concentration for 20 min at room temperature. Cells were then washed with PBS, spun down at 300 g for 5 min, and resuspended in 300 μL of PBS. Cells were run on an Attune NxT™ flow cytometer (Thermo Fisher Scientific) and subsequent analysis was performed using FlowJo software (BD).
Streptococcus pyogenes Cas9 ribonucleoprotein preparation
Ribonucleoproteins (RNPs) were produced by complexing a two-component gRNA to Streptococcus pyogenes Cas9 (SpyCas9). In brief, tracrRNA and crRNA were ordered from Integrated DNA Technologies (IDT), suspended in nuclease-free duplex buffer at 100 μM, and stored in single-use aliquots at −20°C. tracrRNA and crRNA were thawed, and 0.0625 μL of each component was mixed 1:1 by volume and annealed by incubation at room temperature for 5 min to form sgRNA solution for each well of a 96-well plate. Recombinant sNLS-SpCas9-sNLS Cas9 (Aldevron; 10 mg/mL) was added to the complexed gRNA at a 1:1 molar ratio (1000 ng/well, total 0.1 μL) and incubated for 5 min at room temperature to form RNP.
RNP delivery
iPSCs were singularized using Accutase and counted using a Countess® II FL Automated Cell Counter (Thermo Fisher Scientific) with 0.4% Trypan blue viability stain (Thermo Fisher Scientific). iPSCs were then seeded at 15,000 cells/well on 96-well glass-bottom plates (Cellvis) in 100 μL mTeSR1 (WiCell) and 10 μM ROCK inhibitor (Y27632; Selleckchem), 2 days before lipofection or electroporation. On the following day, iPSCs were treated with TSA (0–200 ng/mL) for 16–24 h in optimization experiments described in Supplementary Figure S1, while TSA was applied for 20 h at concentrations of 0, 3.13, 6.25, and 12.5 ng/mL for the rest of the experiments described in the study.
iPSC lipofections were performed using 0.5 μL of Lipofectamine Stem Cell Reagent/well (1000 ng Cas9/well and sgRNA at 1:1 molar ratio). Cells remained undisturbed for 48 h and were then passaged 1:4 using ReLeSR solution followed by daily mTeSR1 media changes for 4 additional days before downstream analysis.
iPSC electroporations were performed using the 4D-Nucleofector System (Lonza) as per the manufacturer's instructions. Briefly, iPSCs were harvested using Accutase (STEMCELL Technologies) and counted. 2 × 105 cells per electroporation were then centrifuged at 300 g for 5 min. Media were aspirated and cells were resuspended using 20 μL of P3 solution (Lonza) with 3 μg of Cas9 and sgRNA at a 1:1 molar ratio. iPSCs were then electroporated using protocol CB-150. After electroporation, samples were incubated in Nucleocuvette at room temperature for 15 min before plating into 6 × 104 cells/well on 96-well glass-bottom plates in mTeSR1 media +10 μM ROCK inhibitor. Media were changed 24 h post-transfection and replaced with mTeSR1 medium daily.
Flow cytometry and fluorescence-activated cell sorting
Flow cytometry was performed on singularized iPSCs using the Attune™ Nxt flow cytometer (Thermo Fisher Scientific) and analyzed using the FlowJo software. Ghost Dye Red 780, ATTO 550, and Green Fluorescent Protein (GFP) fluorescence were detected using 780/60, 585/16, and 530/30 filters in BL1, YL1, and RL3 positions, respectively. Gates were established by running singularized untransfected iPSCs. The percentage of viable cells was calculated as the ratio of Ghost Dye Red 780− cells to the total number of single cells. Transfection efficiency was calculated as the percentage of ATTO 550+ cells to the total number of viable cells on day 2 post-transfection. Gene editing efficiency was calculated as the percentage of GFP− cells to the total number for viable cells on day 6 post-transfection. For the sorting experiments, iPSCs were singularized 6 days post-transfection using Accutase, washed with PBS +20 μM ROCK inhibitor, and centrifuged at 300 g for 5 min.
Ghost Dye Red 780 viability dye (Tonbo Biosciences) was then added at 1:1000 concentration for 20 min at room temperature. Cells were washed again with PBS +20 μM ROCK inhibitor, spun down at 300 g for 5 min, and resuspended in 300 μL of fluorescence-activated cell sorting buffer (PBS +2% BSA +20 μM ROCK inhibitor). GFP− Cells were then sorted into tubes containing mTeSR1 + 20 μM ROCK inhibitor, and seeded onto Matrigel®-coated 6-well polystyrene plates at 100 cells/well to obtain single-cell iPSC clones.
Next-generation sequencing of genomic DNA
DNA was isolated from iPSCs by adding 50 μL of DNA QuickExtract™/well (Epicentre) following treatment by Accutase and centrifugation. The DNA extract solution was incubated at 65°C for 15 min, 68°C for 15 min, and finally 98°C for 10 min. Genomic PCR was performed according to the manufacturer's instructions using Q5 Hot Start polymerase (NEB); primers are listed in Supplementary Table S1. Sequencing indices were added with a second round of PCR using indexing primers (IDT), followed by a purification using the AMPure XP magnetic bead purification kit (Beckman Coulter). Samples were pooled and sequenced on an Illumina® MiniSeq at a run length of 1 × 150 or 2 × 150 bp according to the manufacturer's instructions. Analysis was performed using IDT CRISPR Analysis software, where edited alleles were those that contained indels around the sgRNA target site.
Genome-wide off-target analysis
Genomic DNA from iPSCs was isolated using the Gentra Puregene® Kit (Qiagen) according to the manufacturer's instructions. CHANGE-seq was performed as previously described. 42 Briefly, purified genomic DNA was tagmented with a custom Tn5-transposome to an average length of 400 bp, followed by gap repair with Kapa HiFi HotStart Uracil + DNA Polymerase (KAPA Biosystems) and Taq DNA ligase (NEB). Gap-repaired tagmented DNA was treated with USER™ enzyme (NEB) and T4 polynucleotide kinase (NEB). Intramolecular circularization of the DNA was performed with T4 DNA ligase (NEB) and residual linear DNA was degraded by a cocktail of exonucleases containing Plasmid-Safe ATP-dependent DNase (Lucigen), Lambda exonuclease (NEB) and Exonuclease I (NEB).
In vitro cleavage reactions were performed with 125 ng of exonuclease-treated circularized DNA, 90 nM of SpCas9 protein (NEB), NEB buffer 3.1 (NEB), and 270 nM of sgRNA, in a 50 μL volume. Cleaved products were A-tailed, ligated with a hairpin adaptor (NEB), treated with USER enzyme (NEB), and amplified by PCR with barcoded universal rimers NEBNext® Multiplex Oligos for Illumina (NEB), using Kapa HiFi Polymerase (KAPA Biosystems). Libraries were quantified by qPCR (KAPA Biosystems) and sequenced with 151 bp paired-end reads on an Illumina NextSeq instrument. CHANGE-seq data analyses were performed using open-source CHANGE-seq analysis software. 42
To determine the indel frequency at CHANGE-seq-identified off-target sites, on- and off-target sites were amplified from iPSC genomic DNA obtained using rhAmpSeq system (IDT), with primers listed in Supplementary Table S4, and sequencing libraries were generated according to the manufacturer's instructions. Sequencing was then performed with 150-bp paired-end reads on an Illumina Miniseq instrument. Analysis was performed using CRISPAltRations: IDT rhAmpSeq CRISPR analysis tool.84,85
Antibodies and staining
All cells were fixed for 15 min with 4% paraformaldehyde in PBS (Sigma-Aldrich, St. Louis, MO, USA) and permeabilized with 0.5% Triton™-X (Sigma-Aldrich) for >4 h at room temperature before staining. Hoechst (H1399; Thermo Fisher Scientific, Waltham, MA, USA) was used at 5 μg/mL with a 15-min incubation at room temperature to stain nuclei.
Primary antibodies were applied overnight at 4°C in a blocking buffer of 5% donkey serum (Sigma-Aldrich) at the following concentrations: H3K9Me3 (ab8898; Abcam) 1:500; H3K9Ac (39918; Active Motif) 1:100; H3K27Me3 (C36B11; Cell Signaling Technology) 1:200; and NANOG (AF1997; R&D Systems). Secondary antibodies were obtained from Thermo Fisher Scientific and applied in a blocking buffer of 5% donkey serum for 1 h at room temperature at concentrations of 1:400–1:800. A Nikon Eclipse Ti confocal microscope was used to acquire 100 × images. Images were processed using image analysis software CellProfiler 86 to calculate the chromatin condensation values, 79 as described in Supplementary Figure S2A.
Karyotyping
Cells cultured for at least five passages were grown to 60–80% confluence and shipped for karyotype analysis to WiCell Research Institute, Madison, WI. G-banded karyotyping was performed using standard cytogenetic protocols. 87 Metaphase preparations were digitally captured with Applied Spectral Imaging software and hardware. For each cell line, 20 GTL-banded metaphases were counted, of which a minimum of 5 were analyzed and karyotyped. Results were reported in accordance with guidelines established by the International System for Cytogenetic Nomenclature 2016. 88
Statistical analysis
Unless otherwise specified, p-values were calculated using the nonparametric Kruskal–Wallis test, without assuming normally distributed data, for multiple unmatched comparisons with GraphPad Prism software. Statistical tests were deemed significant at α ≤ 0.05. Technical replicates are defined as distinct wells within an experiment. Biological replicates are experiments performed with different passages of iPSCs. No a priori power calculations were performed.
Results
Optimal TSA treatment
We developed a simple, yet robust, workflow to optimize the drug treatment of iPSCs without extensive use of deep sequencing (Fig. 1A). We utilized a mono-allelic mEGFP-tagged HIST1H2BJ WTC-11 human iPSC line (tag at C-terminus) 89 as it provides twofold advantages. First, targeting the mEGFP locus (referred as HIST1H2B-GFP) allows for easy assessment of iPSC editing efficiency via fluorescence imaging or flow cytometry; the percentage of GFP− cells showed strong linear correlation with the indel percentage determined by deep sequencing of genomic DNA (Supplementary Fig. S1A; R2 = 0.9307). Second, these iPSCs enable in situ live imaging of their cell nuclei to monitor chromatin changes during the process of CRISPR-Cas9 gene editing. We used the Alt-R® CRISPR-Cas9 2-part gRNA system (IDT), which consists of (1) crRNA sequence to target the mEGFP locus in iPSCs (Fig. 1B), and (2) ATTO 550 fluorescent dye-labeled tracrRNA that binds to Cas9.
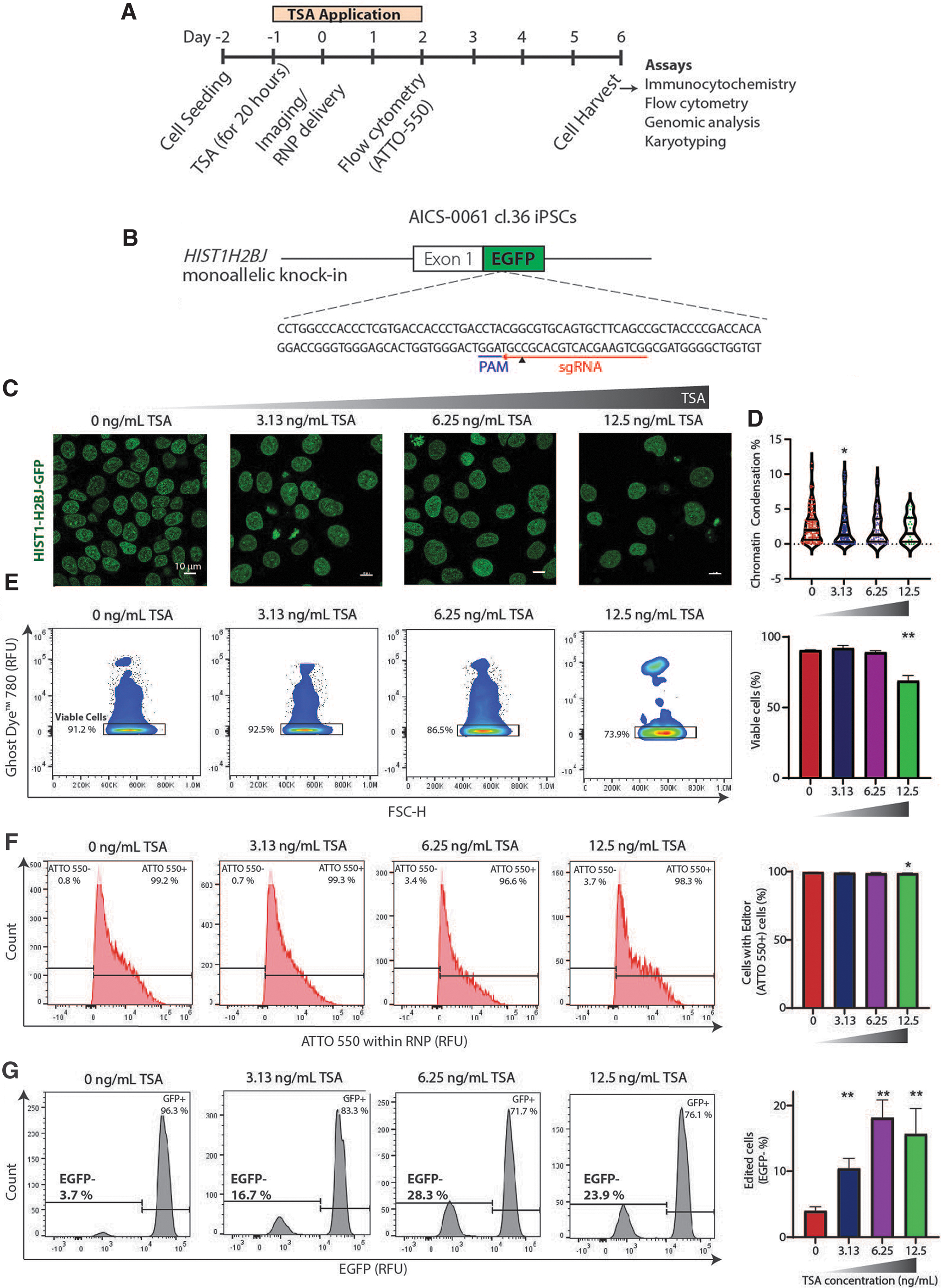
TSA increases CRISPR-Cas9-mediated gene editing efficiency of iPSCs at HIST1H2BJ-GFP locus.
We tested a range of TSA concentrations (0–200 ng/mL) on iPSCs to determine the effect of TSA on cell viability. We observed that cell viability decreased with the increase in TSA concentration (Supplementary Fig. S1B), with a considerable decrease for TSA concentration >25 ng/mL. Thus, we used TSA concentrations of 0, 3.13, 6.25, and 12.5 ng/mL for further studies. Next, we assessed a range of TSA treatment durations (0–24 h) before Cas9 RNP complex delivery and two methods for delivery (lipofection and electroporation) to determine the optimal conditions for gene editing. Since TSA treatment duration of 20 h (Supplementary Fig. S1C) and lipofection (Supplementary Fig. S1D) yielded the highest gene editing efficiencies, we implemented these conditions for our subsequent experiments.
Nuclear imaging of TSA-treated iPSCs
As TSA is known to inhibit HDAC activity on histones, we next sought to examine the effect of TSA treatment on chromatin condensation of iPSCs. We first pretreated the iPSCs with TSA (0, 3.13, 6.25, and 12.5 ng/mL) and subsequently imaged their nuclei. The images were then used as inputs for a CellProfiler software 90 pipeline to output the chromatin condensation% of the nuclei (Supplementary Fig. S2A), defined as the ratio of total heterochromatin intensity to the total nuclear intensity, 79 and is representative of the global chromatin state of cells. Immunofluorescence labeling validated the heterochromatin foci identified in these nuclear images by the CellProfiler pipeline, that is, the H3K9Ac euchromatin histone mark was excluded from the heterochromatin foci, while H3K9Me3 heterochromatin histone mark overlapped with the heterochromatin foci (Supplementary Fig. S2B).
The nuclear image analysis pipeline revealed at least 1% decrease in chromatin condensation upon TSA treatment (Fig. 1C, D), indicating that nuclear imaging is sensitive to the chromatin decondensation induced by TSA. We observed that cell viability as assayed by cell viability Ghost Dye was similar for TSA concentrations up to 6.25 ng/mL and significantly decreases at a TSA concentration of 12.5 ng/mL (Fig. 1E). We then assessed the dose dependence of Cas9 RNP transfection efficiency (% ATTO+ cells/viable cells) by performing flow cytometry analysis on day 2 after Cas9 RNP delivery and noted high transfection efficiencies >95% (Fig. 1F), indicating successful delivery of Cas9 RNP independent of the TSA concentration.
Furthermore, flow cytometry analysis on day 6 after Cas9 RNP delivery revealed a positive correlation between the gene editing efficiency (% GFP− cells/viable cells) and the dose for TSA concentrations of up to 6.25 ng/mL (Fig. 1G). While increasing the concentration of TSA to 6.25 ng/mL showed an ∼3.5-fold increase in gene editing efficiency, an additional increase in the concentration to 12.5 ng/mL did not increase the editing efficiency further. We reasoned that this could be due to the higher cell toxicity as shown by decreased cell viability that occurred at 12.5 ng/mL TSA concentration (Fig. 1E). Taken together, these results indicate that nuclear imaging is sensitive to the chromatin condensation changes induced by TSA and that TSA enhances CRISPR-Cas9-mediated gene editing in a dose-dependent manner.
Deep sequencing analysis of edited iPSCs
After isolating genomic DNA from the treated iPSCs, we performed targeted PCR-amplification around the HIST1H2B-GFP target site in the human genome. Deep sequencing the resulting amplicon library yielded a diversity of reads. Sequencing data were analyzed using the CRISPAltRations tool, which allowed for quantitative analysis of observed indel mutations and their spatial distribution in the HIST1H2B-GFP target region (Fig. 2A). The highest frequency of indels was found three to four nucleotides upstream from the protospacer adjacent motif (PAM) sequence, consistent with reports of type II CRISPR systems.91,92
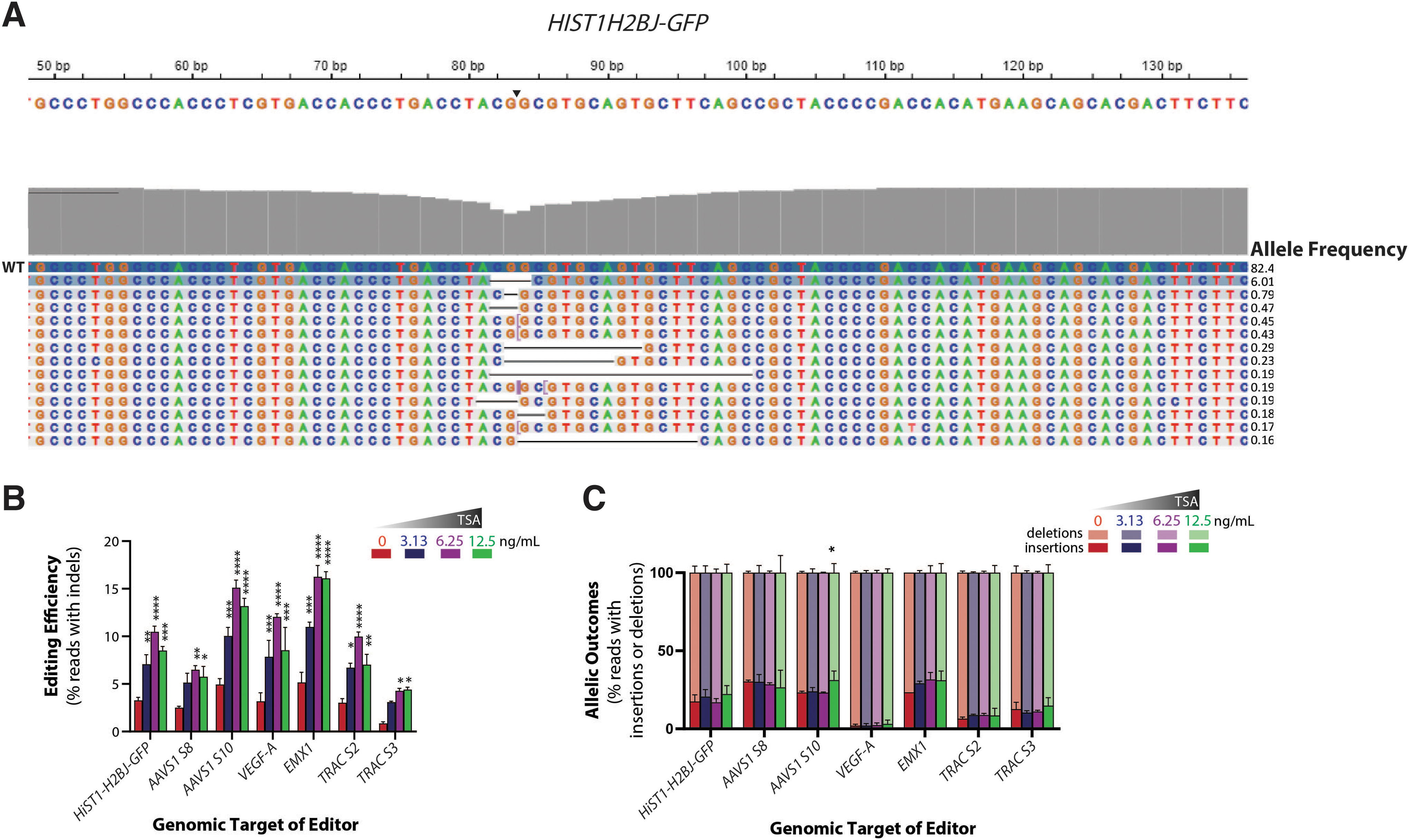
TSA increases on-target gene editing efficiency at several endogenous genes in iPSCs.
Multiple gRNA sequences targeting different sites of open (HIST1H2BJ and AAVS1) and closed chromatin (TRAC, VEGFA, EMX1) were selected to investigate if the observed TSA-induced increase in gene-editing efficiency at HIST1H2BJ-GFP locus could be extended to other endogenous genes with variable chromatin states (Supplementary Table S1). For each of these edited targets, we performed deep sequencing on the edited cells obtained after TSA treatment. We harvested the genomic DNA on day 6 after Cas9 RNP delivery. PCR using primers flanking the sgRNA target sites was then performed and prepared for next-generation sequencing (NGS) using the Illumina MiniSeq™ system.
TSA treatment increased gene-editing efficiency at all the sites independent of the gRNA sequence and initial chromatin state of the loci (Fig. 2B). Across all the sites, the percentage of edited reads containing insertion events and deletion events did not undergo significant change (Fig. 2C) upon TSA treatment, and the insertion and deletion profiles around the cut site were largely similar (Supplementary Fig. S3A, B; Supplementary Table S2). The edited reads contained higher deletion events (∼50–80%) than insertion events (∼5–20%). Greater deletion events relative to insertion events is a common outcome in deep sequencing analyses of human cells edited by SpyCas9.91,93 Taken together, these results indicate that TSA treatment increases the frequency of indels but is unlikely to change the overall mutation spectrum at the target site.
Off-target profiling and karyotypic analysis of edited iPSCs
Because undesired off-target editing could be promoted by TSA treatment, we performed a highly sensitive genome-wide, off-target assay for our TSA-based editing strategy by CHANGE-seq. 85 This analysis yielded a frequency distribution of the potential off-target sites for the HIST1H2B-GFP gRNA (Fig. 3A, B). Twelve sites scattered across the genome (Supplementary Table S3) were nominated as the top off-target sites and amplified from genomic DNA using the rhAmpSeq system84,94 followed by deep sequencing (Supplementary Table S4). Using the CRISPAltRations tool, 84 we tracked the reads containing indels (Supplementary Table S5) at each of the nominated off-target sites. No significant changes in these off-target modifications were found upon TSA treatment at HIST1H2B-GFP off-target sites (Fig. 3C).

Off-target analysis of edited iPSCs.
Furthermore, we found that the highest normalized on-target to off-target edit ratio was obtained for a TSA concentration of 6.25 ng/mL, which we consider to be the optimal TSA concentration for generating gene-edited iPSCs. To broaden our analysis to two other targets, we additionally assessed 10 top off-targets at AAVS1 Site 10 and top 7 off-target sites at EMX1 obtained from previously reported CHANGE-seq 49 and GUIDE-seq 95 assays, respectively. This analysis showed no significant increase in indel% at off-target sites and the highest normalized on-target edit ratio at a TSA concentration of 6.25 ng/mL (Fig. 3D, E), which is similar to the HIST1H2B-GFP site.
Apart from off-target modifications, edited iPSCs can lose pluripotency and have genomic abnormalities, including translocations and even undergo chromothripsis. 96 We assessed the pluripotency of iPSC clones that underwent gene editing at the HIST1H2BJ-GFP locus from different TSA treatment conditions by assessing the levels of a key pluripotency transcription factor, NANOG 97 (Fig. 4A). The edited iPSC clones were negative for GFP indicating that they have been edited and positive for NANOG indicating that they retain pluripotency after editing using TSA. Next, we isolated multiple iPSC clones that underwent gene editing at the HIST1H2B-GFP locus from the 6.25 ng/mL TSA treatment condition and expanded them for subsequent characterization to check for any genomic abnormalities. Karyotyping analysis indicated no genomic abnormalities in four of the five clones, while one clone showed an interstitial duplication in the long (q) arm of chromosome 20 in 5 of the 20 cells examined (Fig. 4B).

Pluripotency marker and karyotypic analysis of TSA-treated edited iPSC lines.
This genetic variant is a known recurrent acquired duplication at this location in human pluripotent stem cell cultures and did not consistently arise in all our edited lines.
Discussion
Relative to the traditional CRISPR-Cas9 gene editing workflow, our workflow with optimal TSA treatment can increase the gene-editing efficiency up to twofold to fourfold without increased off-target mutations, while maintaining genomic integrity and pluripotency.
Optimal TSA treatment surprisingly increases the precision of the editing, as demonstrated in the greater on- to off-target ratio at three different target sites (Fig. 3). There is a boost in editing efficiency at the on-target site without increasing the off-target potential of the editor. The kinetics of SpyCas9 RNP binding at off-target sites are known to be different from those at the on-target site,98–100 and thus, strong target strand recognition at the on-target site may be involved in the mechanisms by which TSA increases precision. No increase in detectable off-target editing could indicate that the sequence specificity of the gRNA could be more important than the chromatin state of the off-target site. Furthermore, the kinetics of DNA repair processes may be different for the reduced level of DNA double-strand break or nick formation at the off-target site.101,102
These repair processes may also be affected by TSA differently at the on-target site versus the various off-target sites in the genome that undergo less DNA double-strand breaks or nicks. However, given the large conservation of mutational spectrum at most of the sites evaluated over all TSA treatments (Fig. 2; Supplementary Fig. S3), it is unlikely that TSA is globally switching end-joining DNA repair pathways. Importantly, we note that our PCR-based rhAMP-seq amplification methods have limited sensitivity at around 1% of reads, and that proportional changes for off-target modification at <1% were not able to be observed in this study. Future studies with more sensitive genomic profiling at off-target sites may provide deeper insights into the mechanisms involved with increased precision upon TSA treatment.
Our workflow is easy to implement and could be combined with the in situ assessment of cell state. We demonstrate that chromatin decondensation upon TSA treatment is observable, and this observation could be combined with the in situ assessment of other biological processes—such as cell proliferation, apoptosis, and DNA damage via imaging of GFP, CellEvent Caspase 3/7 Detection reagent, 103 and immunostained histone H2AX phosphorylation 104 —to enable the identification iPSCs more amenable to gene editing. In situ monitoring of one or more of these processes in future work could ultimately reduce the cost and times associated with culturing and passaging to isolate rare edited iPSC clones.
Overall, we describe an easy small-molecule method with well-established CRISPR-Cas9 editors to manufacture gene-edited iPSCs in an efficient and high-quality manner.
Footnotes
Acknowledgments
We thank members of the Saha laboratory for helpful discussion and comments on the article. We thank the University of Wisconsin (UW) Carbone Cancer Center Flow Cytometry Laboratory for assistance with flow cytometry experiments, and Aldevron for technical support with Cas9 proteins. We thank Tyler Duellman and the University of Wisconsin-Madison Biotechnology Center Gene Expression Center for providing single nuclei library preparation and next-generation sequencing services. We thank Branimir Bugarija and Gavin Kurgan for help with designing and analyzing rhAmpSeq panels. We also thank Allen Institute for Cell Science for providing the GFP-reporter iPSCs. We thank Nikolai Fedorchak for assistance with confocal imaging.
Authors' Contributions
Conceptualization: K.M. and K.S. Experimental design: K.M. and K.S. Methodology: K.M., N.K., and C.R.L. Data analysis: K.M. and N.K. Supervision: K.S. and S.Q.T. Writing—original draft: K.M. Writing—review and editing: K.M., K.S., and N.K.
Author Disclosure Statement
K.M. and K.S. have filed a patent application on this work. The remaining authors declare no competing financial interests.
Funding Information
This work was supported by the National Science Foundation [CBET-1350178]; National Institutes of Health [R35 GM119644]; University of Wisconsin-Madison Stem Cell and Regenerative Medicine Center Graduate Student Fellowship to K.M.; NIH NHGRI training grant to the Genomic Sciences Training Program [5T32HG002760 to N.K.]; Wisconsin Institute for Discovery; Wisconsin Alumni Research Foundation. National Institutes of Health (NIH) Office Of The Director (OD) Somatic Cell Genome Editing (SCGE) initiative grant [U01AI157189 to S.Q.T.].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.