
Abstract
Rhodopsin (RHO) mutations such as Pro23His are the leading cause of dominantly inherited retinitis pigmentosa in North America. As with other dominant retinal dystrophies, these mutations lead to production of a toxic protein product, and treatment will require knockdown of the mutant allele. The purpose of this study was to develop a CRISPR-Cas9-mediated transcriptional repression strategy using catalytically inactive Staphylococcus aureus Cas9 (dCas9) fused to the Krüppel-associated box (KRAB) transcriptional repressor domain. Using a reporter construct carrying green fluorescent protein (GFP) cloned downstream of the RHO promoter fragment (nucleotides −1403 to +73), we demonstrate a ∼74–84% reduction in RHO promoter activity in RHOpCRISPRi-treated versus plasmid-only controls. After subretinal transduction of human retinal explants and transgenic Pro23His mutant pigs, significant knockdown of rhodopsin protein was achieved. Suppression of mutant transgene in vivo was associated with a reduction in endoplasmic reticulum (ER) stress and apoptosis markers and preservation of photoreceptor cell layer thickness.
Introduction
In a survey of 1000 consecutive families clinically diagnosed with an inherited retinal disease, we identified a pathological genotype in 76% of them. 1 Of these, ∼12% carried disease-causing dominantly inherited variants, of which 36.5% were in the gene encoding rhodopsin (RHO). The most common disease-causing mutation in patients with RHO-associated retinitis pigmentosa (RP) is a gain-of-function missense variant at amino acid residue 23 (Pro23His). 2
This proline-to-histidine substitution creates a misfolded protein that aggregates within the cell and induces an unfolded protein response (UPR), endoplasmic reticulum (ER) stress, and photoreceptor cell death. 3 Unlike recessive disorders, in which delivery of a wild-type copy of the gene can provide rescue from further degeneration, 4 treatment of dominantly inherited RHO-associated RP will likely require knockdown or “silencing” of the mutant gene product before gene augmentation.
Several technologies, including hammerhead ribozymes (hhRzs),5–9 RNA interference (RNAi),10–13 and antisense oligonucleotides (ASOs), 14 each of which have specific advantages and disadvantages, have been evaluated as treatments for dominantly inherited disease. Viral delivery of hhRZs and RNAi slowed photoreceptor degeneration and achieved functional rescue, respectively, in Pro23His transgenic rodent models.5,15 Rho repression and functional rescue were also demonstrated in a Pro23His transgenic rat model treated with ASOs. 14 However, these technologies rely on interaction with target RNA that can be inaccessible due to molecular folding, protein coating, and structural fluctuations.9,16,17
The recently developed CRISPRi system is a promising alternative for targeted gene repression. This approach relies on the use of a catalytically inactive Cas9 (dCas9) protein, generated by making single amino acid substitutions in each of its two nuclease domains: D10A in the RuvC domain and H840A in the HNH domain of Streptococcus pyogenes Cas9; or D10A and N580A in the Staphylococcus aureus Cas9 RuvC and HNH domains, respectively.18,19 When fused to the repressive chromatin modifier domain Krüppel-associated box (KRAB) and paired with a guide targeted to a gene promoter, this CRISPR-dCas9–KRAB (i.e., CRISPRi) system can be used to attenuate gene expression. 20
In this study, CRISPRi constructs targeted to the promotor region of RHO were developed. The ability to suppress rhodopsin expression was demonstrated in both a reporter cell line and human retinal explants in vitro. To evaluate CRISPRi-mediated transcriptional repression and treatment efficacy in vivo, subretinal injections of adeno-associated virus serotype 5 (AAV5)-packaged CRISPRi constructs were performed in a transgenic pig model of retinal degeneration harboring human Pro23His rhodopsin.21,22 After transduction, we detected reduced expression of human mutant rhodopsin, which correlated with outer nuclear layer (ONL) preservation in eyes treated with CRISPRi expressing vectors compared with contralateral vehicle controls. This study provides a paradigm to develop CRISPRi therapies for RHO-associated RP and other dominantly inherited retinal dystrophies.
Materials and Methods
Plasmid constructs
Adeno-associated virus transgene cassette plasmids
Guides targeting each of the regulatory elements RET1, NRE, and the TATA box, as well as the transcription start site of the human RHO promoter, were designed using the Benchling platform (www.benchling.com) (Table 1). Oligonucleotides were synthesized (Integrated DNA Technologies, Coralville, IA), annealed, phosphorylated, and cloned into the BsaI sites of an adeno-associated virus (AAV) transgene cassette plasmid. This plasmid had AAV2 ITRs flanking a bicistronic expression cassette containing (1) the human U6 promoter and single guide sequences upstream of (2) the cytomegalovirus promoter-sequence upstream of (3) the catalytically inactive S. aureus Cas9 (dSaCas9) CDS fused to (4) the KRAB transcriptional repressor domain sequence (pAAV-hU6-sgRNA-CMV-dSaCas9–KRAB).
Guide sequences used to target the human RHO promoter
RHO, rhodopsin.
RHOp-copGFP reporter plasmid
The GFP reporter plasmid carried the cytomegalovirus (CMV) immediate early enhancer and ∼1.3 kb RHO regulatory sequence 23 upstream of the Pontellina plumata copGFP CDS. AAV2 ITRs flanked the expression cassette (pAAV-RHOp-copGFP).
Standard curve plasmids used in quantitative polymerase chain reaction (qPCR)
The copGFP sequence was amplified from the pGreenZeo plasmid (Systems Biosciences, Palo Alto, CA) and TA cloned into pCR2.1-TOPO-TA vector (Thermo Fisher Scientific, Waltham, MA) using the primers 5′-CATGCCCGCCATGAAGA-3′ and 5′-CAGATGGCTGGCAACTAGAA-3′. The 18S sequence was amplified and TA cloned into pCR2.1-TOPO-TA using the primers 5′-CATTCGAACGTCTGCCCTATC-3′ and GCCCTTCCGTCAATTCCTTTA-3′.
CRISPR-dSaCas9 functional screen in human cells
Transfection
pAAV-hU6-sgRNA-CMV-dSaCas9–KRAB and pAAV-RHOp-copGFP reporter plasmids (500 ng each) were cotransfected into HEK293T cells using Lipofectamine 3000 (Thermo Fisher Scientific) according to manufacturer's instructions.
RNA isolation and complementary DNA synthesis
Forty-eight hours post-transfection total RNA was isolated using the NucleoSpin RNA kit (Takara Bio, San Jose, CA). RNA was DNase treated using RQ1 RNase-Free DNase according to manufacturer's instructions (Promega Corporation, Madison, WI). DNase-treated RNA (200 ng) was used as template in complementary DNA (cDNA) synthesis reactions using the Hi Capacity cDNA Synthesis Kit (Thermo Fisher Scientific) following the manufacturer's protocol.
qPCR
Total number of copGFP transcripts from three individual transfection experiments were quantified using SYBR Green reagents on the QuantStudio 3 platform (Thermo Fisher Scientific). copGFP transcripts were amplified using the primers 5′-GCTACGGCTTCTACCACTTC-3′ and 5′-ATGCGGGTGTTGGTGTAG-3′. 18S transcripts were amplified using the primers 5′-CTGAGAAACGGCTACCACATC-3′ and 5′-GCCTCGAAAGAGTCCTGTATTG-3′. cDNA (100 ng) from each sample was amplified in triplicate.
Standard curves (109–103 copies/μL) were generated and evaluated in triplicate and visualized through scatter plot with the logarithmic y-axis scale plotted against the cycle threshold (Cq) on the x-axis. An exponential trend line was used to determine values by which to determine number of copies using the equation: y = (multiplier)e(-integer)x, where y = number of copies and x = Cq.
Number of copGFP copies was normalized to number of 18S copies in each sample and GFP transcript reduction was calculated as the percentage of number of copGFP copies/number of 18S copies ratios in controls treated with reporter plasmid alone. Primer efficiencies were determined by linear regression analysis using the equation: efficiency = (10(−1/slope) − 1) × 100. copGFP and 18S primer efficiencies were 92.18% ± 0.26% and 97.87% ± 1.59%, respectively.
AAV5 vector production
Recombinant AAV5 vectors (hereafter referred to as AAV5–RHOpCRISPRi) were produced by triple plasmid transfection of HEK293T cells. Cells were harvested at 48 h post-transfection, lysed by homogenization, and purified through iodixanol gradient and anion exchange column. The eluate was concentrated and subjected to buffer exchange with rAAV storage buffer (water for injection, 180 mM NaCl, 10 mM Na3PO4, pH ∼7.0). Viral genomes per milliliter were determined through qPCR.24–26
Transduction of human retinal explants
Human donor eyes were obtained from the Iowa Lions Eye Bank (Coralville, IA) within 4–6 h of death (Table 2) and prepared for explant culture as previously described. 27 Then 10 μL AAV5–RHOpCRISPRi (1 × 1012 vg/mL) was applied directly beneath explants and 10 μL was added to the culture medium beneath transwell inserts (20 μL vector per well). Untransduced explants were cultured as controls. Human explants were cultured for 7 days in the presence of virus.
Human eye donors
Genome-wide RNA-seq analysis of off-target gene suppression in AAV5–RHOpCRISPRi transduced human retinal explants
RNA was isolated from cultured donor retina samples as described above. Sequencing libraries were prepared by the Genomics Division of the Iowa Institute of Human Genetics. The TruSeq Stranded messenger RNA (mRNA) library kit was used (Illumina). Libraries were pooled and sequenced on the NovaSeq6000 instrument (Illumina) generating 100 bp paired-end reads. An index for the human transcriptome was generated from the human genome assembly GRCh38 using Salmon's index function (Version 1.10.1). 28
FASTQ quality was confirmed using FastQC (Version 0.11.9) and reads from samples passing initially quality control were quantified against the above index using Salmon's quant function (Version 1.10.1) with the following settings:—validateMappings—seqBias—gcBias. Downstream analysis was carried out using the DESseq2 package (Version 1.38.3) 29 in R (Version 4.2.2). Data were filtered to only include transcript IDs with at least eight counts (i.e., a mean of two counts per sample). Differential expression analysis was carried out using the DESeq function with default parameters.
Transcripts were considered as differentially expressed if their log2 fold change value was >1 (i.e., twofold) and their adjusted p-value was <0.05. Two pairwise comparisons were made: between donors and between treatment groups. Data were visualized using the EnhancedVolcano package (Version 1.16.0) in R. The raw and processed data are available on Gene Expression Omnibus (GEO) under the accession number GSE244786.
AAV5 vector injection into subretinal space of Pro23His RHO mutant swine
All procedures were approved by the institutional animal care and use committee of the University of Iowa (IACUC No. 7051070) and were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Subretinal injections
AAV5–RHOpCRISPRi (300 μL) vector was delivered into the subretinal space of one eye in each of eight 4-week-old Pro23His transgenic swine21,22 (four males and four females; two males and two females per each of two time points) by a single experienced pig surgeon (E.H.S.). Control buffer injections were administered contralaterally. To control for experimenter bias, the surgical staff and scientist performing the postsurgical analysis were masked to the treatment condition. Conditions were unmasked after study completion.
Each animal underwent pars plana vitrectomy under general anesthesia using a 23-gauge instrument platform (Alcon, Fort Worth, TX) after sterile preparation was done, including induction of posterior vitreous detachment. A 41-gauge flexible polyamide cannula (MedOne, Sarasota, FL) was used to create a small retinotomy and inject 300 μL of virus (1 × 1011 vg) into the subretinal space manually with optimized parameters that have been previously described.30,31 Sclerotomies were sutured and each eye was rinsed in 5% povidone–iodine.
Two and 12 weeks postinjection, eyes were enucleated and a 20-mm section encompassing the injection site was dissected using a biopsy punch and flash frozen before protein isolation and sectioning. The surgeons and researchers performing the postharvest data collection were masked to treatment condition.
Ophthalmoscopic examinations
Indirect ophthalmoscopic examinations were performed at the time of sacrifice and compared with the fundus drawings recorded at the time of injection. Examinations were conducted by board-certified ophthalmologists who had completed vitreoretinal fellowships (E.H.S., S.R.R., and I.C.H.). Particular attention was paid at sacrifice to identify any treatment-induced ocular changes. Animals were anesthetized using ketamine (4–6 mg/kg), dexmedetomidine (0.02–0.04 mg/kg), and butorphanol (0.3 mg/kg) through IM.
Once sedated, isoflurane inhalation was administered through mask followed by endotracheal tube placement. Eyes were dilated with tropicamide (1% v/v) and phenylephrine (2.5% v/v). Artificial tears ointment was applied to eyes to prevent drying of the corneas. Meloxicam (0.4 mg/kg) or ketoprofen (1–3 mg/kg) was administered as needed for pain relief.
Isolation of retinal protein and Western blotting
Human retinal explants (N = 3 independently cultured and virus-treated explants) were homogenized in fresh RIPA buffer (Thermo Fisher Scientific) supplemented with cOmplete™ protease inhibitor cocktail (MilliporeSigma) and lysates cleared through tabletop microcentrifugation. Supernatant protein concentrations were determined using a BCA kit according to the manufacturer's instructions (Pierce, Rockford, IL).
Western blots were performed as described previously.26,32,33 Fifty micrograms of total protein per sample was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 4–20% Tris-glycine gels; Thermo Fisher Scientific), transferred to PVDF using an iBlot 2 dry blotting system (Thermo Fisher Scientific), blocked in 3% bovine serum albumin in 1 × phosphate buffered saline (PBS) supplemented with 0.1% Tween-20, and probed with primary antibodies overnight at 4°C.
Blots were probed with the following primary antibodies: mouse antirhodopsin (Cat. No. MABN15; 1:500 dilution; Millipore Sigma, Burlington, MA), rabbit antirecoverin (RCVRN; Cat. No. AB5585; 1:2000 dilution; Millipore Sigma), and mouse anti-GAPDH (Cat. No. ab8245; 1:2500 dilution; Abcam, Cambridge, United Kingdom) as a loading control. The following secondary antibodies were used: goat antimouse (Cat. Nos. 32430; 1:5000; Thermo Fisher Scientific) and goat antirabbit (Cat. Nos. 31460; 1:5000; Thermo Fisher Scientific) cross-adsorbed secondary antibodies conjugated to horseradish peroxidase.
Antigen–antibody complexes were visualized on X-ray film through enhanced chemiluminescence using SuperSignal West Pico PLUS (Thermo Fisher Scientific). Blots were stripped using Restore Western Blot Stripping Buffer (Thermo Fisher Scientific). Densitometry was quantified using FIJI (National Institutes of Health, Bethesda, MD) software. 34
Two and 12 weeks post-transduction of Pro23His transgenic swine, eyes (N = 4 treated and 4 vehicle controls from each timepoint) were enucleated and the injection area was excised using a 10 mm biopsy punch that captured the neural retina, underlying retinal pigmented epithelium, choroid, and sclera. Excised injection areas were bisected and half of each was fixed, cryoprotected, and sectioned as previously described and as below. 27
The remaining half of bisected retinae of Pro23His transgenic animals were used in Western blot analyses as described above and probed with the following primary antibodies: mouse antirhodopsin (Cat. No. MABN15; 1:500 dilution; Millipore Sigma), rabbit antirecoverin (Cat. No. AB5585; 1:2000; Millipore Sigma), rabbit anti-PERK (Cat. No. 5683; 1:500; Cell Signaling Technology), rabbit anti-BiP (Cat. No. 3177; 1:500; Cell Signaling Technology), rabbit anti-CHOP (Cat. No. 5554; 1:500; Cell Signaling Technology), rabbit anticleaved PARP (Cat. No. 5625; 1:500; Cell Signaling Technology), rabbit anticleaved caspase 3 (Cat. No. 9579; 1:500; Cell Signaling Technology), and mouse anti-GAPDH (Cat. No. ab8245; 1:2500; Abcam) as a loading control.
Immunohistochemistry and confocal imaging
After harvest, retinal explants were rinsed, fixed, mounted in Optical Coherence Tomography (OCT), and sectioned as previously described.27,33,35 Sections were blocked in SuperBlock™ (PBS) blocking buffer (Thermo Fisher Scientific) and labeled with the primary mouse antirhodopsin antibody (Cat. No. MAB5316; 1:100; EMD Millipore) and secondary goat antimouse AlexaFluor 488 conjugated antibody (Cat. Nos. A11001; 1:1000; Thermo Fisher Scientific). Nuclei were counterstained with DAPI (Cat. No. D9542; 1:2000; Sigma-Aldrich).
Labeled sections were imaged using Leica TCS SPE DMi8 inverted confocal microscope system (Leica Microsystems, Wetzlar, Germany). Transgenic pig retinal sections were imaged as described above with rabbit antirecoverin (Cat. Nos. MA1-932; 1:100; Thermo Fisher Scientific) and goat antirabbit AlexaFluor conjugated 647 antibody (Cat. Nos. A21245; 1:1000; Thermo Fisher Scientific). Phenotypic analyses were performed with primary mouse antirhodopsin antibody as indicated above and AlexaFluor conjugated 568 lectin peanut agglutinin (PNA; Cat. No. L-32458; 1:500; Molecular Probes).
Outernuclear layer thickness measurements
Recoverin and DAPI labeled images of retinal sections from untreated control and AAV5–RHOpCRISPRi-treated transgenic pig eyes (N = 4 eyes per condition) were masked to the observer. Six columns of DAPI-stained nuclei from each section (N = 12 sections per eye) extending from the outer plexiform layer to the RPE cell layer were counted and the average number of nuclei per column were calculated for each eye.
Digital polymerase chain reaction-mediated number of copies analysis
As the mutant Pro23His rhodopsin pig was originally generated through Somatic Cell Nuclear Transfer (SCNT), 22 in addition to two copies of wild-type pig rhodopsin, it also expresses an unknown number of copies of human mutant rhodopsin. To accurately determine the number of copies of mutant human Pro23His rhodopsin in the pig, digital polymerase chain reaction (PCR) using probes targeting human rhodopsin (unknown number of copies) and the pig gene RNASEH1 (i.e., two copies per cell) was performed. Genomic DNA was isolated from four transgenic pigs (two males and two females) using the NucleoSpin Tissue Kit (Takara Bio) according to manufacturer's instructions.
The Sus scrofa genome size (2.5 × 109 bp) was used to determine genome mass through the Thermo Fisher DNA calculator tool (www.thermofisher.com/DNA-calculator). Then 1 × 104 genome copies were used as template in multiplexed digital PCRs using Absolute Q Digital PCR Master Mix (Thermo Fisher) and primer/probes targeting human RHO (F—CTGAGCTGAGGCTCAAAGAA, R—CAAACATGGCCCGAGATAGA probe—HEX/TTCCAACTCAACTCTGCACCCGTC/ZEN/IowaBlack™ FQ) and pig RNASEH1 (F—GGATGTAGTTACCGTGGTCAAG, R—GGTGCACACACAGTCAGATAA, probe—HEX/TGACTCCACCACCAGCACTGTTC/ZEN/IowaBlack FQ; Integrated DNA Technologies).
Reactions were run on the Applied Biosystems QuantStudio Absolute Q Digital PCR machine (Thermo Fisher Scientific) and analyzed with QuantStudio Absolute Q Digital PCR Software (version 1.3.1) according to manufacturer's protocol. Automatic thresholds were used for each reaction.
Statistical analyses
Data were analyzed using GraphPad Prism (v9; GraphPad Software, La Jolla, CA). All values were presented as mean plus or minus standard error of the mean.
Results
RHOpCRISPRi plasmid reagents reduce rhodopsin expression in human cells
To screen guide RNA efficacy in reducing RHO expression, we designed CRISPRi plasmids carrying a single guide RNA (sgRNA) targeting the human RHO promoter and S. aureus dCas9 (dSaCas9) fused to the KRAB transcriptional repressor domain (Fig. 1A). To test the efficiency of our engineered plasmids, a reporter plasmid carrying GFP driven by the CMV immediate early enhancer and ∼1.3 kb of upstream RHO regulatory sequence 23 was delivered to HEK293T cells with one of four engineered CRISPRi constructs targeting the RHO promoter (RHOpCRISPRi) (Fig. 1B).
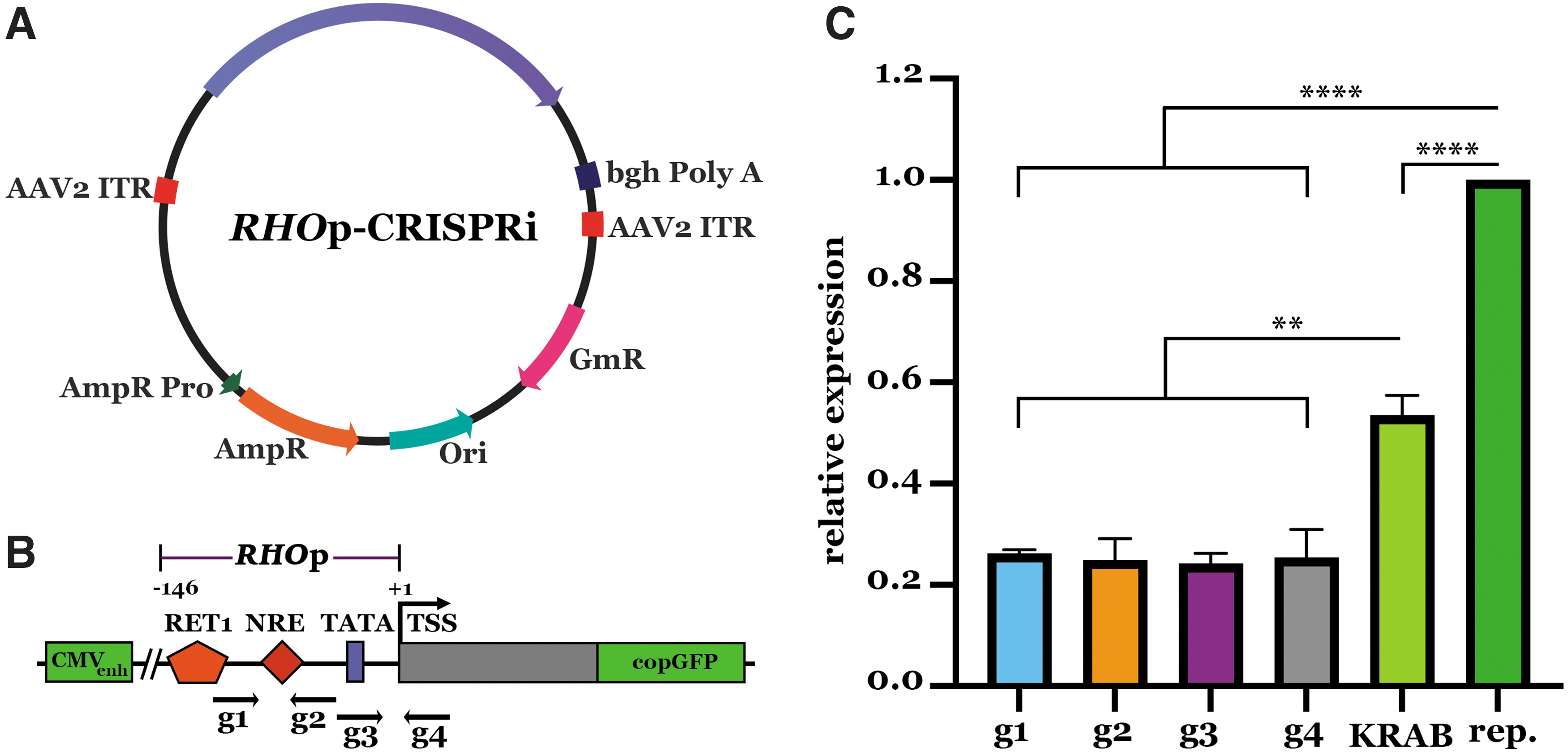
CRISPRi-based transcriptional repression of RHO promoter.
Forty-eight hours after transfection, we isolated total RNA and quantified GFP mRNA expression using real-time PCR. As controls, we transfected cells with the reporter plasmid alone or with a plasmid expressing only dSaCas9–KRAB (i.e., CRISPRi plasmid lacking RHOp sgRNA). Cells treated with RHOpCRISPRi and reporter plasmids demonstrated a 74–76% reduction in total GFP expression when compared with control cells treated with reporter plasmid only (Fig. 1C). Notably, qPCR assays in cells treated with reporter plasmid and a plasmid expressing only dSaCas9–KRAB, which lacked the targeting sgRNA sequence, also demonstrated a significant reduction in GFP reporter expression when compared with reporter-only controls.
As reported previously, these observations indicate some indiscriminate transcriptional inhibition from the repressor domain. 18 Regardless, RHO-specific targeting plasmids significantly reduced GFP reporter expression beyond that of the dSaCas9–KRAB control (Fig. 1C). As the degree of reduction induced by each of the engineered RHOpCRISPRi constructs was virtually identical, we chose the plasmid containing g1 (which had the least amount of variability in repeat experiments) for packaging into AAV vectors and subsequent experiments.
AAV5–RHOpCRISPRi reduces RHO expression in human retina in vitro
To evaluate the ability of our AAV5–RHOpCRISPRi vector to target and reduce endogenous RHO expression in human retina, we employed an ex vivo organotypic retinal explant model as previously described. 27 We and others have shown that AAV5 vectors transduce human photoreceptor cells with high efficiency.27,36 Therefore, g1-expressing plasmids were packaged into AAV5 vectors using standard triple transfection protocols.37,38 Human retinal explants were transduced with AAV5–RHOpCRISPRi at an estimated multiplicity of infection of 104 vector genomes per cell.
Untransduced retinal explants from each donor served as controls. At 7 days post-transduction, dCas9–KRAB expression was evaluated using rtPCR, and RHO expression was evaluated through immunohistochemistry and Western blot analyses. Expression of dCas9–KRAB transcript was detected in retina transduced with AAV5–RHOpCRISPRi from all three donors (Fig. 2A). Western blot analyses on whole protein lysates revealed immunoreactive bands corresponding to RHO monomer (35 kDa), dimer (70 kDa), and trimer (110 kDa) (Fig. 2B).

CRISPRi machinery successfully targets and decreases rhodopsin protein expression in human retinal explants.
Equal levels of RCVRN and the loading control GAPDH were detected (Fig. 2A). Densitometric analysis of RHO normalized to the loading control GAPDH revealed a significant reduction in mean RHO abundance in retinal explant cultures transduced with the AAV5–RHOpCRISPRi vector as compared with control (i.e., 0.96 ± 0.45 vs. 2.19 ± 0.75, respectively) (Fig. 2B). These studies demonstrate the ability of the developed AAV5–RHOpCRISPRi vector to target and reduce endogenous rhodopsin expression in human retina in vitro.
As indicated in Figure 1, expression of dSaCas9–KRAB in the absence of targeting sgRNA sequence caused a reduction in GFP reporter expression as compared with reporter-only controls. As this finding indicates that indiscriminate transcriptional inhibition from the repressor domain is possible, off-target suppression of retinal transcripts was evaluated genome wide through RNAseq analysis. Only 22 genes were determined to be differentially expressed between CRISPRi transduced and control retina (Fig. 3A, B). Interestingly, a far greater number of transcripts were found to be differentially expressed between tissue donors (Fig. 3A, C). These findings indicate that transduction of human retina with AAV5–RHOpCRISPRi does not induce widespread off-target suppression of retinal transcripts.
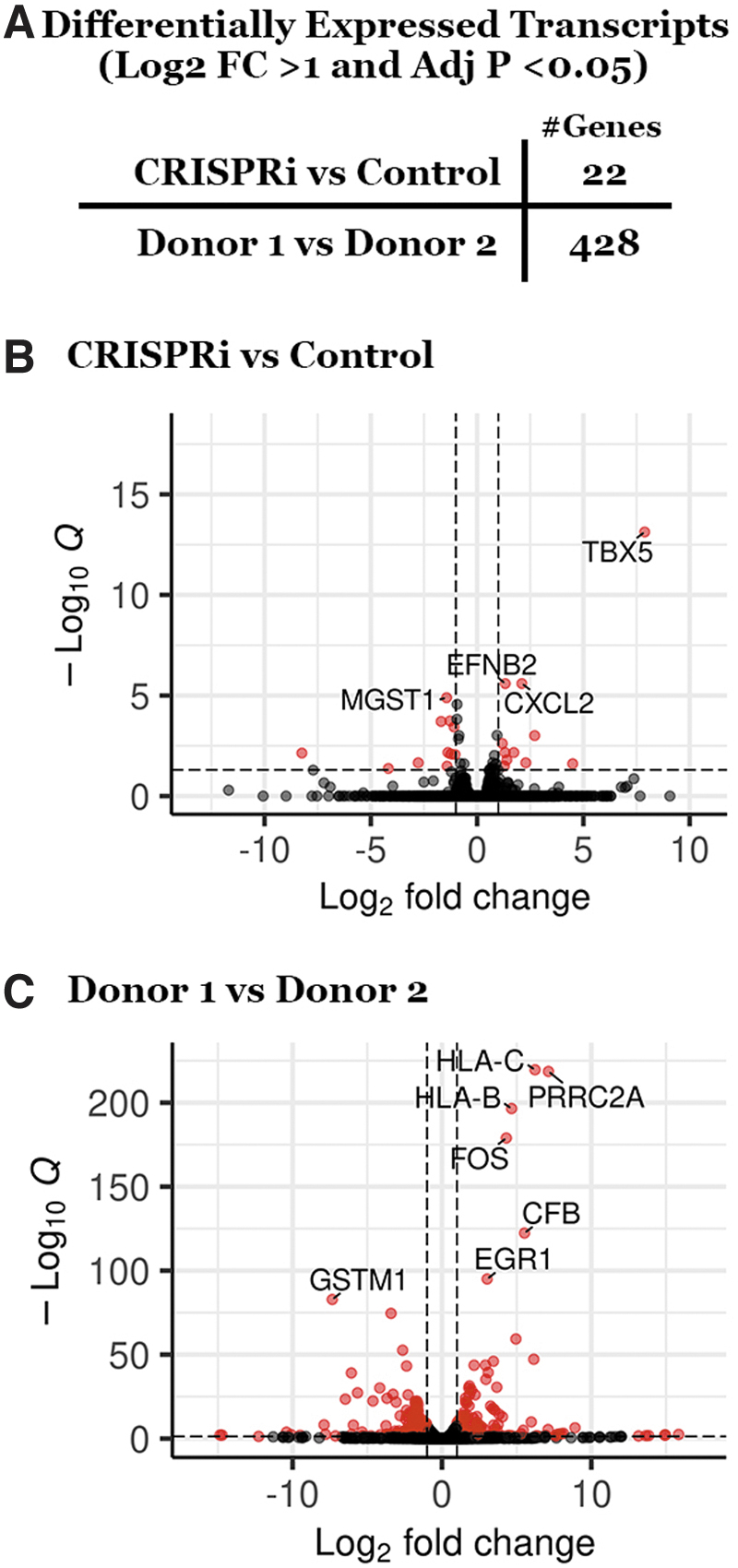
Genome-wide off-target transcript suppression analysis.
AAV5–RHOpCRISPRi reduces RHO expression in vivo
To demonstrate the feasibility of AAV5–RHOpCRISPRi delivery in vivo, we extended our studies to Pro23His transgenic swine, which expresses both human Pro23His mutant RHO (Fig. 4A) and wild-type swine RHO.21,22 This model is ideal for these studies as (1) the human mutant transgene can be selectively targeted leaving the wild-type swine gene intact and (2) the model undergoes rapid panretinal photoreceptor degeneration allowing early detection of treatment effect (Fig. 4B–D).
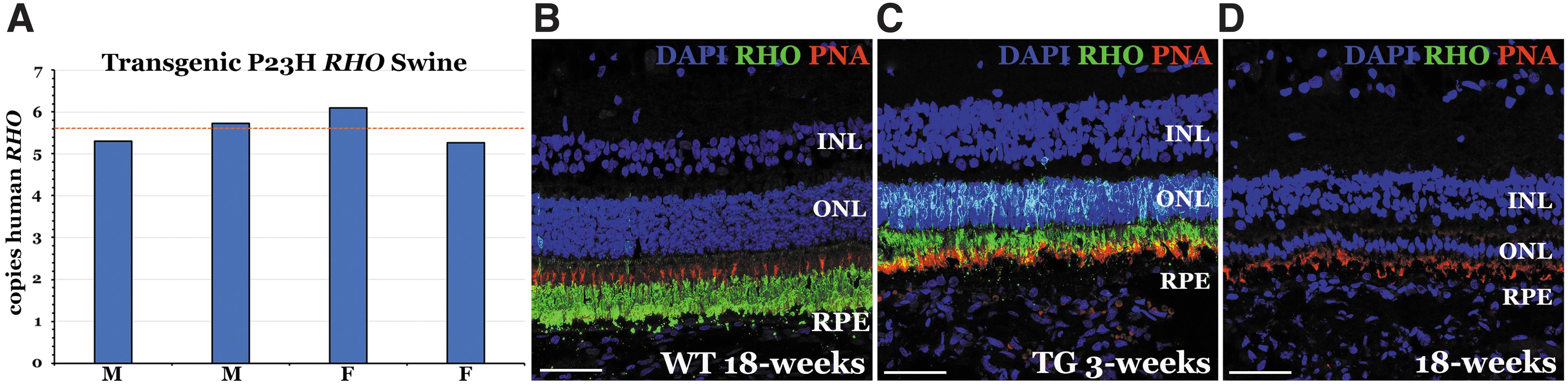
Number of transgene copies and phenotypic analysis of the Pro23His rhodopsin transgenic pig model.
To determine whether suppression of human mutant Pro23His RHO and retention of the wild-type gene was able to slow disease progression, the AAV5–RHOpCRISPRi vector (1 × 1011 vector genomes) was injected into the subretinal space of one eye of each transgenic animal, and vehicle was injected contralaterally (N = 4, 2F and 2M). At 2 weeks postinjection, Western blot analyses revealed reduced expression of all three human RHO isoforms in AAV5–RHOpCRISPRi-transduced eyes as compared with contralateral vehicle controls and with unchanged levels of RCVRN (Fig. 5A).
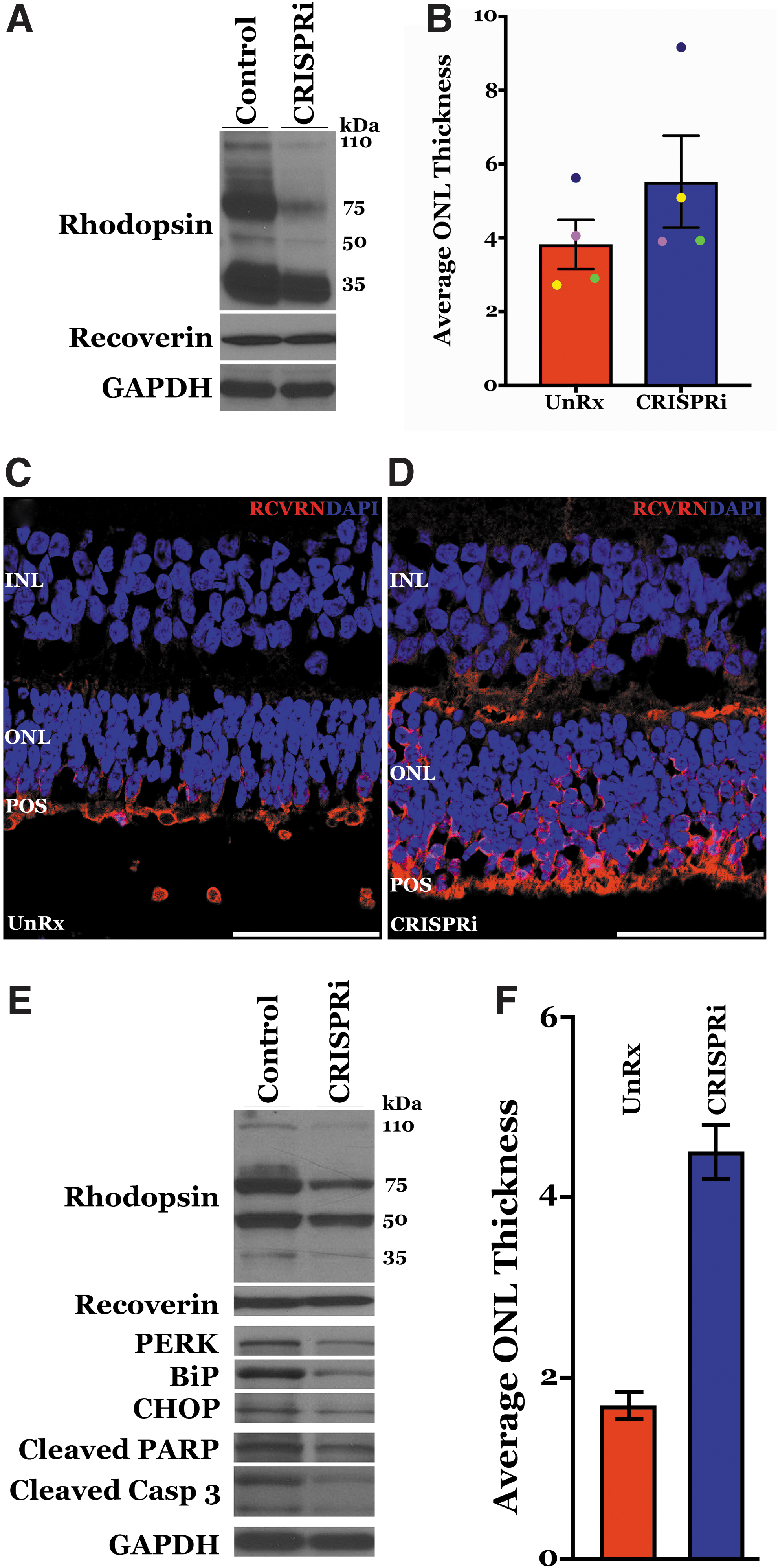
AAV5–RHOpCRISPRi subretinal delivery mitigates endoplasmic reticulum (ER) stress and preserves ONL thickness in Pro23His transgenic swine model.
To determine whether suppression of human Pro23His RHO mitigated disease progression, ONL thickness (measured as average number of DAPI-stained nuclei per column) was determined through immunohistochemistry and confocal microscopy using an antibody directed against recoverin. At 2 weeks postinjection, counting of DAPI-labeled nuclei in images of AAV5–RHOpCRISPRi-transduced retinae revealed a mean ONL thickness of ∼1.5 nuclei greater than in vehicle control-treated retinae (N = 4; 5.52 ± 0.80 nuclei in the treated retinae vs. 3.83 ± 0.50 nuclei in controls, respectively) (Fig. 5B–D).
To determine whether this treatment effect persisted, we evaluated RHO expression in a separate cohort of animals (N = 4) at 12 weeks postinjection. We detected reduced RHO expression in the AAV5–RHOpCRISPRi-treated eye versus the contralateral control in one of the four animals (Fig. 5E and Supplementary Fig. S2). As the proline-to-histidine amino acid substitution at position 23 of rhodopsin is known to cause photoreceptor cell death through ER stress, 3 we assessed expression of the ER stress markers PERK, BiP, and CHOP and the apoptosis markers cleaved PARP and cleaved caspase 9 in this 12-week post-treatment animal.
Expression of all ER stress and cell death markers evaluated was reduced in the AAV5–RHOpCRISPRi-treated eye relative to the contralateral vehicle control eye (Fig. 5E). Reduced expression of human mutant rhodopsin; the ER stress markers PERK, BiP, and CHOP; and the apoptosis markers cleaved PARP and cleaved Caspase 9 correlated with increased ONL thickness in the treated versus control eye of this animal (4.5 ± 0.3 nuclei in the RHOpCRISPRi-treated eye vs. 1.7 ± 0.2 nuclei in the vehicle control) (Fig. 5F).
Discussion
Mutations in the RHO gene, the most common of which is Pro23His, are the leading cause of dominantly inherited RP in the United States. 1 Pro23His rhodopsin causes photoreceptor cell death and loss of vision through production of misfolded protein, induction of the UPR, and ER stress. 3 Although AAV-mediated gene augmentation has shown promise for the treatment of recessive and X-linked disorders,4,39–45 dominantly inherited diseases in which the mutation creates a toxic protein product are not likely to benefit from gene augmentation strategies alone. For diseases such as this, an approach that reduces expression of the mutant allele may be beneficial.
Here we describe studies employing CRISPR-Cas9-mediated transcriptional repression that attempted to prevent mutant rhodopsin-induced photoreceptor cell death. We created CRISPR-Cas9 transcriptional knockdown reagents using catalytically inactive S. aureus Cas9 (dCas9) fused to the KRAB transcriptional repressor domain and sgRNAs targeted to the promoter region of the human RHO gene, which we refer to as RHOpCRISPRi.
Using a reporter construct carrying GFP cloned downstream of the RHO promoter fragment (nucleotides −1403 to +73), we demonstrate efficient repression of rhodopsin promoter-driven GFP expression. After packaging into an AAV5 vector, we were able to demonstrate the ability of our engineered RHOpCRISPRi construct to suppress endogenous rhodopsin expression in a human retinal explant model in vitro. At 2 weeks after subretinal injection in Pro23His rhodopsin mutant pigs in vivo, rhodopsin expression was reduced, and photoreceptor cell layer thickness was preserved in AAV5–RHOpCRISPRi-treated eyes versus contralateral vehicle controls.
Interestingly, despite receiving the same dose of virus and being treated at the same age, the treatment effect was more pronounced in some animals than in others. Differences in treatment effect were even more striking as survival times were increased. Specifically, of the four Pro23His pigs treated in the 12-week survival group, one was found to have a sustained reduction in rhodopsin expression, which was associated with increased ONL thickness and reduced expression of both ER stress (i.e., CHOP, BiP, and PERK) and apoptosis markers (i.e., cleaved caspase 9 and cleaved PARP).
To prevent rod photoreceptor cell death and noncell-autonomous disease progression (i.e., secondary cone cell loss), a minimum number of target cells must be transduced. 46 Although this threshold may vary from one form of inherited retinal degeneration to the next, we know that treatment effect is proportional to both disease state and the number of cells transduced. In general, the likelihood of achieving a sustained treatment effect rises as the treatment age decreases (i.e., less cell death) and the number of photoreceptor cells transduced increases.
As all Pro23His pigs in this study were of the same age at time of treatment and the rate of disease progression is virtually identical between animals, the most plausible explanation for the difference in treatment effect is differences in transduction efficiency. As the transgenic pigs used in this study were derived through artificial insemination of an outbreeding sow (i.e., offspring are genetically diverse), differences in AAV transduction efficiency from one animal to the next were not unexpected. In fact, we previously demonstrated using a human retinal explant model that genetic diversity between donors results in significant variability in AAV transduction efficiency, 27 validating the use of animal models that incorporate genetic diversity, as results are ostensibly more translatable to human health.
One of the advantages of the Pro23His mutant rhodopsin pig model used in this study is that the human mutant rhodopsin transgene is expressed independent of wild-type pig rhodopsin. This made it possible to specifically target and repress the mutant transgene without altering wild-type expression. Although excellent for demonstrating a treatment effect in vivo, gene repression is unlikely to be effective as a stand-alone therapy. Specifically, as the targeted rhodopsin promoter is expected to be nearly identical for both affected and unaffected (Pro23) alleles, repression of the mutant allele (His23) only is unlikely.
For instance, to achieve allele-specific repression, we attempted to target an single nucleotide polymorphism (SNP) in the 5′ UTR (untranslated region) of RHO (rs 7984) using our in vitro GFP reporter assay. Unfortunately, no difference in GFP transcript repression was observed (i.e., a single base pair change was insufficient to confer allele selectivity) (Supplementary Fig. S1). For the CRISPRi approach described here to be useful, concurrent gene augmentation will likely be required. Careful design of the gene augmentation vector will be important, as photoreceptors have been shown to be sensitive to high levels of RHO expression.47–49
To prevent overexpression toxicity, ideally RHO would be delivered under the control of its endogenous promoter. Unfortunately, doing so would allow the CRISPRi reagents developed here to target both the endogenous mutant allele and the therapeutic transgene. As such, selection of a heterologous promoter, such as that of the rhodopsin kinase gene (GRK1), will likely be required.
Although the CRISPRi approach presented here is promising, achieving complete suppression of mutant rhodopsin while expressing the wild-type gene at the appropriate level may be challenging. As such, exploration of alternative gene independent strategies designed to prevent secondary cone photoreceptor cell loss may be useful. As rod photoreceptor cells are the primary consumer of choroidal oxygen, their loss during disease progression results in elevated oxygen levels in the subretinal space.50–52 When coupled with free radical generation associated with the normal visual cycle, cone photoreceptor cells are subject to significant oxidative stress, which eventually becomes too great to withstand. 50
AAV-mediated delivery of genes such as PGC1α and NRF2, transcription factors that globally regulate antioxidant defense, has been shown to significantly enhance cone cell survival in the face of rod cell death in several mouse models of RP.53,54 To mitigate rhodopsin-associated disease most effectively, a combinatorial approach may be ideal. Specifically, a dual knockdown and overexpression strategy targeting rod photoreceptor cells combined with an antioxidant approach targeting cones may provide the most sustainable treatment effect possible.
Conclusions
In summary, we present data demonstrating successful production of novel CRISPRi reagents capable of suppressing mutant rhodopsin expression, mitigating ER stress and slowing the rate of rod photoreceptor cell degeneration in a large animal model of disease. A combination strategy involving CRISPRi-mediated gene suppression with traditional gene augmentation may enable the treatment of dominantly inherited retinal degeneration. However, further work is still needed to achieve a sustained treatment effect in relevant model systems toward eventual human clinical trials.
Footnotes
Authors' Contributions
E.R.B. contributed to conceptualization, methodology, validation, formal analysis, investigation, writing, and visualization. L.A.W. was involved in methodology, validation, formal analysis, investigation, writing, and visualization. N.K.M. took charge of methodology, validation, formal analysis, investigation, writing, and visualization. M.K.A. carried out methodology and writing—review and editing. M.J.L., C.M.C., and C.J. were involved in investigation.
S.R.R., E.H.S., and I.C.H. were involved in investigation and writing—review and editing. J.W.R. carried out methodology, resources, and writing—review and editing. E.M.S. and R.F.M. oversaw resources, writing, supervision, and funding acquisition. B.A.T. was in charge of conceptualization, resources, writing, visualization, supervision, project administration, and funding acquisition.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported, in part, by the University of Iowa Institute for Vision Research and the National Eye Institute R01 EY026008, P30 EY025580.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.