
Abstract
Rhodnius prolixus is currently the model vector of choice for studying Chagas disease transmission, a debilitating disease caused by Trypanosoma cruzi parasites. However, transgenesis and gene editing protocols to advance the field are still lacking. Here, we tested protocols for the maternal delivery of CRISPR-Cas9 (clustered regularly spaced palindromic repeats/Cas-9 associated) elements to developing R. prolixus oocytes and strategies for the identification of insertions and deletions (indels) in target loci of resulting gene-edited generation zero (G0) nymphs. We demonstrate successful gene editing of the eye color markers Rp-scarlet and Rp-white, and the cuticle color marker Rp-yellow, with highest effectiveness obtained using Receptor-Mediated Ovary Transduction of Cargo (ReMOT Control) with the ovary-targeting BtKV ligand. These results provide proof of concepts for generating somatic mutations in R. prolixus and potentially for generating germ line-edited lines in triatomines, laying the foundation for gene editing protocols that could lead to the development of novel control strategies for vectors of Chagas disease.
Introduction
Chagas disease is a neglected tropical disease that inflicts great health and socioeconomic impacts in the Americas. Caused by the protozoan parasite Trypanosoma cruzi, reduviid insects represent one of the most common transmission routes, either by the insects delivering parasites during blood feeding or by oral ingestion of the parasite-carrying insects with açai fruit. 1 Chagas disease treatments are limited, however, and vector control is essential to restrict spread of the disease.
The kissing bug Rhodnius prolixus is the most well-studied laboratory species among the triatomines that transmit T. cruzi. With a sequenced genome, 2 gene identification and transcriptome analyses have revealed important aspects of R. prolixus physiology and evolution.3–6 Gene knockdown by parental RNA interference (pRNAi) is effective in R. prolixus, allowing investigation of gene function.7–9 Such studies would be greatly enhanced, however, by advances in transgenic and gene-editing technologies, which could enable efficient functional genetic studies and potentially lead to the development of novel biological control strategies for the insect vectors.
CRISPR-Cas9 (clustered regularly spaced palindromic repeats/Cas-9 associated) methods have revolutionized precise genome editing in a broad range of animal lineages. These effective and widely used tools benefit from its simplicity and the great number of putative target sequences for practically every gene. Adapted from bacteria, the most common technique involves complexing the Cas9 endonuclease with single guide RNAs (sgRNAs), which recognize target sequences in the genome and direct site-specific double-stranded breaks (DSBs) by the action of the Cas9 nuclease. 10 These DSBs are then corrected by a series of repair mechanisms, such as Non-Homologous End-Joining (NHEJ) or Homology Directed Repair (HDR). 11 NHEJ is error-prone and often generates short insertions and deletions (indels) at the cut site, leading to loss-of-function mutations. HDR, on the other hand, enables the precise insertion of exogenous sequences at the cut site, permitting facile site-directed modification of target sequences and insertion of transgenic gene cassettes.
Most animal CRISPR protocols rely on the injection of sgRNA/Cas9 complexes or plasmids containing sequences for the production of CRISPR elements into the embryo by embryonic microinjection. These injections are performed during early embryonic development when nuclei are easily accessible, increasing the chance of gene edition in the germ line. Experiments of this type are quite challenging to perform in triatomines such as R. prolixus. First, oogenesis in these animals is strictly dependent on blood ingestion. 12 Three to five days after blood feeding, vitellogenic oocytes start to develop and females lay ∼30–40 eggs in the next 3 weeks. Therefore, collecting embryos at the appropriate stage for injections (0–2 h development) requires a large number of ovipositing animals. Second, eggs are encapsulated by a hard chorion 13 that is extremely difficult to penetrate for injection with DNA constructs, resulting in low viability of injected embryos.
Alternative methodologies that bypass the need for embryonic microinjections have been developed, which involves injecting CRISPR components into the adult female hemolymph followed by uptake of these components by developing oocytes.14–18 While such methods do not yet allow the incorporation of exogenous sequences into target sites by HDR, they have been shown to generate small deletions and base alterations by NHEJ that may produce effective gene knockouts.
Previously, we identified visible markers that would serve as appropriate target sites for gene editing in R. prolixus. Through RNA interference we identified several loci that when disrupted lead to visible cuticle or eye color phenotypes with little or no effect on insect fertility or viability. Such viable and fertile phenotypes are essential for generating edited lines and following the heritability of editing events through multiple generations. 19
Here we tested two techniques: Receptor-Mediated Ovary transduction of Cargo (ReMOT Control 14 ) and Direct Parental CRISPR (DIPA CRISPR 17 ) for delivery of Cas9/gRNA (guide RNA) complexes to oocytes and generation of gene edited offspring. We show that while DIPA-CRISPR was not effective in R. prolixus, ReMOT control was able to generate CRISPR-edited generation zero (G0) offspring for several targeted cuticle and eye color genes. These results are the first to accomplish gene editing in triatomine bugs, opening possibilities for the development of germ-line gene editing strategies to address functional genetic questions in triatomines and applied strategies for Chagas disease control.
Methods
Insect rearing
R. prolixus rearing was performed at 28°C and 70–75% humidity, with animals fed on rabbit blood throughout all developmental stages. Animal care and experimental protocols were conducted following guidelines of the Committee for Evaluation of Animal Use for Research from the Federal University of Rio de Janeiro (CEUA-UFRJ, No. 01200.001568/2013-87, order No. 155/13). Technicians dedicated to the animal facility at the Institute of Medical Biochemistry (UFRJ) conducted all aspects related to rabbit husbandry under strict guidelines to ensure careful and consistent animal handling.
Injections to determine ovary intake of P2C- and BtKV-tagged fluorescent proteins
Two tags have been used to signal for the uptake of proteins into vitellogenic oocytes: P2C, from Drosophila melanogaster yolk protein, 14 and BtKV, from Bemisia tabaci vitellogenin. 16 To determine whether P2C sequences and BtKV sequences were efficient to deliver proteins to R. prolixus oocytes, we injected young adult females with untagged mCherry, P2C-mCherry, and BtKV-mCherry into the abdomen (1 μL, 2,5 or 5 μg/μL). As control for background fluorescence, we injected females with the same buffer used for mCherry proteins. Tagged and untagged mCherry protein was produced as in Chaverra-Rodriguez et al. 14 Subsequently, animals were blood fed and 2–3 days later, the ovaries were dissected, fixed in paraformaldehyde 4%, mounted in 70% glycerol and observed under a Leica MZ10F Fluorescent Stereomicroscope.
To test whether P2C and BtKV were efficient for oocyte delivery in other Chagas disease vectors, we obtained Triatoma infestans animals from the International Reference laboratory for Triatomine Taxonomy at Fundação Oswaldo Cruz, Rio de Janeiro, injected the females and analyzed the oocytes as above. To determine the amount of fluorescence uptake, we used the analyze function in Fiji, collecting the mean gray value for a constant area in vitellogenic oocytes, since mature chorionated oocytes display a great amount of background fluorescence.
sgRNA design and synthesis
sgRNAs were designed against Rp-yellow (Rp-y), Rp-white (Rp-w), Rp-scarlet (Rp-sca), and Rp-aaNAT loci to target sequences at the beginning of the coding region or close to enzyme activation sites using CHOP CHOP. 20 sgRNAs were either synthesized at Synthego or synthesized in vitro with Invitrogen Megascript™ T7 Transcription (AM1334), 21 using the primers in Supplementary Tables S1 and S2.
gRNA/Cas9 injections
gRNA/Cas9 complexes were prepared as in Ref 14. 14 Briefly, R. prolixus females were starved and injected with a solution containing 5 μg Cas9 and 2.5 μg of gRNA. Cas9 proteins used were P2C-Cas914 and BtKV-Cas9, 16 prepared as above, or commercially purchased Cas9 (PNABIO). The injection was performed between the third thoracic and the first abdominal segment, using a 10 μL Hamilton microsyringe adjusted so as not to exceed 2 μL of solution for each adult female. The microsyringe was cleaned before and after each injection with hot water, 70% alcohol, and finally milliQ water, to avoid possible contamination of the animals during the procedure. After the injection, the insects were allowed to recover for 1 day and then fed using rabbit blood. Females were observed for 15 days to obtain information on survival and egg laying. Injections were performed prior blood feeding, since those performed after blood feeding resulted in a great number of female deaths.
In vitro cleavage assays
To ensure the effectiveness of designed gRNAs, we performed in vitro cleavage assays (Supplementary Fig. S2) with gRNA/Cas9 complexes as in Martin-Martin et al. 21 Briefly, the ability of each sgRNA to direct Cas9 cleavage of the target DNA was tested in vitro by checking the integrity of the target DNA region after incubation of polymerase chain reaction (PCR) amplified and purified target sequences (with Monarch® PCR & DNA Cleanup Kit T1030), sgRNA (30 ng; 3.2 μM) and Cas9 protein (400 ng; 3.8 μM). After incubation and Cas9 inactivation, the DNA was analyzed by electrophoresis to search for the presence of fragments resulting from cleavage of the double-stranded DNA.
Quantification of fecundity and embryonic viability
Eggs were collected from 2 to 3 weeks after blood ingestion, counted, and the hatch rate defined after 20 days at 28°C. To define possible effects of gRNA/Cas9 injections on fecundity, in some experiments, females were placed in individual vials for egg collection. Embryonic viability was defined by counting the number of hatched embryos per total number of eggs laid.
Phenotypic screening
Injected females were reared individually (particularly for multiple sgRNA injections) or in groups of four to five females. All first instar nymphs (>48 h after hatching) were screened for cuticle or eye color phenotypes using a dissecting scope, under a stream of CO2. Animals were fed and kept to develop throughout all stages in an attempt to cross the animals displaying a visible phenotype. However, none of the animals displaying visible phenotypes survived the 6 months required to reach adulthood. A subset of G1 brothers and sisters of the animals presenting visible phenotypes were sequenced to check for putative edition events in heterozygosity.
Image acquisition
Images displaying visible phenotypes were obtained using a Leica MZ10F Stereomicroscope, with animals sleeping under a stream of CO2.
Genomic DNA extraction and PCR protocol
Genomic DNA was extracted from G0 nymphs using a modification of the single-fly method developed for D. melanogaster. 22 Briefly, DNA was extracted from single first nymphs or nymph exuvia by macerating in a 50 μL solution containing 49 μL of “squishing buffer” (10 mM Tris-Cl pH 8.0; 1 mM EDTA; 25 mM NaCl) and 1 μL of proteinase K (20 mg/mL). After maceration in solution, an incubation period is carried out at 37°C for 20 min for enzymatic action, followed by 5 min at 95°C for enzyme inactivation. At the end of the process, the liquid part was transferred to a new tube to discard the exoskeleton debris. The DNA-containing solution was kept at −20°C.
Target sequences were PCR amplified using primers flanking the gRNA target site (Supplementary Table S3) and either sent for Sanger or for Illumina sequencing as detailed below. For animals presenting visible phenotypes PCR products were also cloned into TOPO TA cloning vector (Invitrogen) for transforming NEB 5-alpha Competent Cells (Escherichia coli C2987P). At least 10 colonies per condition were analyzed. Plasmid DNA was isolated from each colony under standard protocols and purified with QIAprep Spin Miniprep Kit (Cat. No./ID: 27104) for Sanger sequencing.
Sanger sequencing
PCR products or plasmid DNA were used for Sanger sequencing at the genomic DNA sequencing facility at the Institute of Biophysics Carlos Chagas Filho at UFRJ. Sequencing results were aligned to reference sequences using Snapgene. At least 10 wild-type animals were used to sequence around each gRNA target site to enable the detection of putative single nucleotide polymorphism (SNPs).
Amplicon deep sequencing
To identify and quantify mutations at the target loci, sequences around the gRNA cleavage site were amplified by PCR from G0 or G1 nymph genomic DNA using Q5 Hot-Start High Fidelity Polymerase. PCR products were purified, and 20 μg/μL of each sample was sent for Illumina paired-end Amplicon-based deep sequencing at Genewiz (Amplicon-EZ; https://www.genewiz.com/Public/Services/Next-Generation-Sequencing/Amplicon-Sequencing-Services/Amplicon-EZ/?&). All resulting reads were analyzed using CRISPResso2 (https://crispresso.pinellolab.partners.org/submission) with wild-type sequences as reference. Primer sequences and amplicon sizes for each target are detailed in Supplementary Table S3.
Statistical analyses
Statistical analyses were performed through unpaired t test with Welch's correction. Differences were considered significant at p < 0.05. All statistical analyses were performed using the Prism 7.0 software (GraphPad Software).
Results
ReMOT delivery of proteins to triatomine ooctyes
We tested whether ReMOT Control could deliver components to triatome oocytes in vivo by first injecting 4–7 days postecdysis adult R. prolixus females with mCherry protein fused to either the P2C- or the BtKV-oocyte delivery sequences,14,16 and measured the amount of fluorescence in developing oocytes. Unmodified mCherry was injected as a control. The P2C peptide is derived from the N-terminal portion of D. melanogaster Yolk Protein 1 and has been shown to increase the delivery of sgRNA/Cas9 complexes to oocytes in multiple Dipteran, Hymenopteran, Coleopteran, and Hemipteran species.15,23,24 The BtKV peptide is derived from the B. tabaci vitellogenin binding sequence and has been shown to deliver cargo to vitellogenic oocytes in the hemipteran B. tabaci. 16
Following injections into R. prolixus oocytes, we observed that unmodified mCherry could be taken up to some extent when injected into the hemolymph, but that oocyte uptake levels were significantly increased when mCherry was fused to either the P2C or BtKV ovary targeting sequences. Furthermore, we found that the BtKV peptide was more effective than P2C based on quantitation of fluorescence intensity in the ovaries (Fig. 1). To test the generality of these observations, we also injected untagged and tagged forms of mCherry into female T. infestans (another important vector of Chagas disease). We observed similar results as those in R. prolixus (Fig. 1F–H), indicating that P2C- and BtKV increase oocyte delivery of molecular cargo in multiple triatomine vectors.
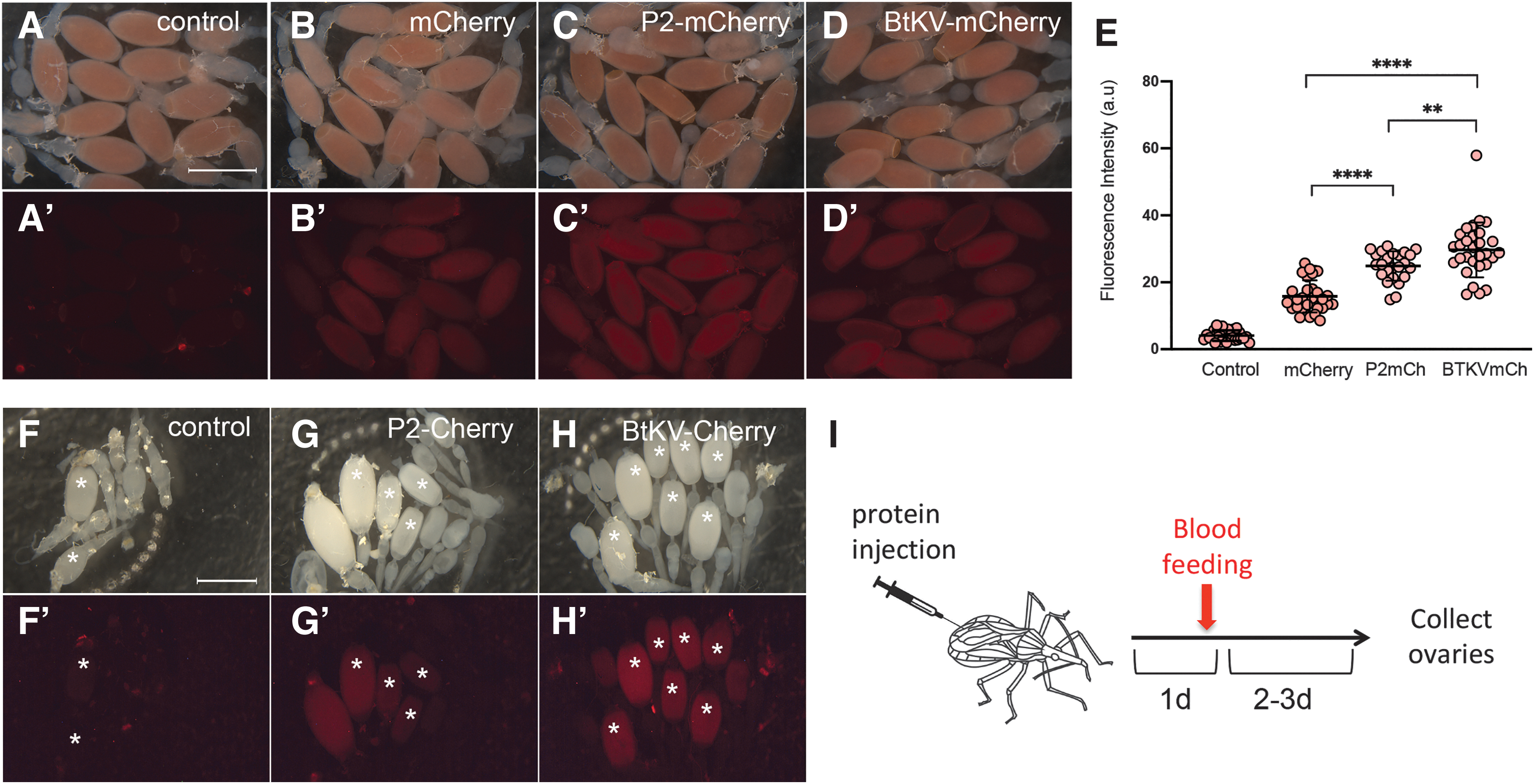
ReMOT tagged proteins are efficiently transferred to triatomine vitellogenic oocytes.
CRISPR-Cas9 editing for genes encoding visible markers in R. prolixus
Next, we tested the conditions for gene editing in R. prolixus, using maternal delivery of sgRNA/Cas9 RNP complexes. We designed sgRNAs targeting coding sequences of genes that encode cuticle or eye pigmentation proteins, expecting that loss-of-function (lof) mutations in these genes should produce visible phenotypes. 19 The targeted loci were Rp-yellow (Rp-y), which encodes an enzyme required for the synthesis of melanin during cuticle pigmentation; Rp-scarlet (Rp-sca), which encodes an ABC transporter that transports pigments into the eye; Rp-white (Rp-w), which encodes a second ABC transporter that transports pigments into the eye; and Rp-aaNAT, which encodes an enzyme required for cuticle pigmentation (Supplementary Table S1). sgRNAs designed for the different loci were injected as preformed ribonucleoprotein complexes with either unmodified Cas9 (DIPA-CRISPR), 17 or with P2C-Cas914 or BtKV-Cas916 to test ReMOT Control. Most injections included the endosomal escape reagent chloroquine as it has been shown to increase ReMOT effectiveness in some systems (Supplementary Fig. S1). Injections performed before blood feeding have no significant effect on survival or on the number of eggs laid per female. 19
Among all injections performed, we were able to identify animals displaying visible phenotypes in G0 nymphs for complexes targeting the Rp-y, Rp-sca, and Rp-w loci, but observed no obvious phenotypes for the Rp-aaNAT locus (Table 1). In contrast, no visible phenotypes were observed for any of these loci in nymphs following DIPA-CRISPR injections. Among the animals injected with the ReMOT control method, we observed several offspring displaying red eyes (instead of the wild-type black eye phenotype) when mothers were injected with Rp-sca gRNA, one individual displaying white eyes derived from a mother injected with Rp-w gRNA, and one individual (y29) displaying a yellow cuticle obtained from a mother injected with Rp-y gRNA. For Rp-sca, the eye phenotype was seen either by injecting a single sgRNA for Rp-sca or when coinjected with other gRNAs. Total DNA prepared from a subset of G0 nymphs that displayed no visible phenotypes was subjected to PCR for amplification of the regions surrounding the sgRNA target site followed by Sanger sequencing. No gene editing events were detected by this method. Nymphs with visible phenotypes were followed each day in an attempt to maintain the animals for genetic crosses as adults. However, these animals were among the nymphs that did not survive to the next stage, and DNA was extracted from these individuals upon death and prepared for DNA sequence analysis.
CRISPR editing efficiency in G0
P2C-Cas9 injections were performed in the absence of chloroquine. All other injections were performed with 0.5 mM chloroquine.
Sequencing was performed by Sanger using total genomic DNA.
Sequencing was performed by Illumina Deep Sequencing and/or Sanger Sequencing using amplified sequences cloned into bacteria.
Eggs counted for 1 week only (second week after blood feeding), total nymphs screened exceeded 300 for these conditions.
A mixture of four gRNAS were injected with Cas-9 protein: white 1, yellow 2, aaNAT 1, scarlet 2.
DIPA, Direct Parental; G0, generation zero; gRNA, guide RNA; nd, not determined, NA, Not applicable.
Confirmation of gene editing events in Rp-yellow, Rp-white, and Rp-sca
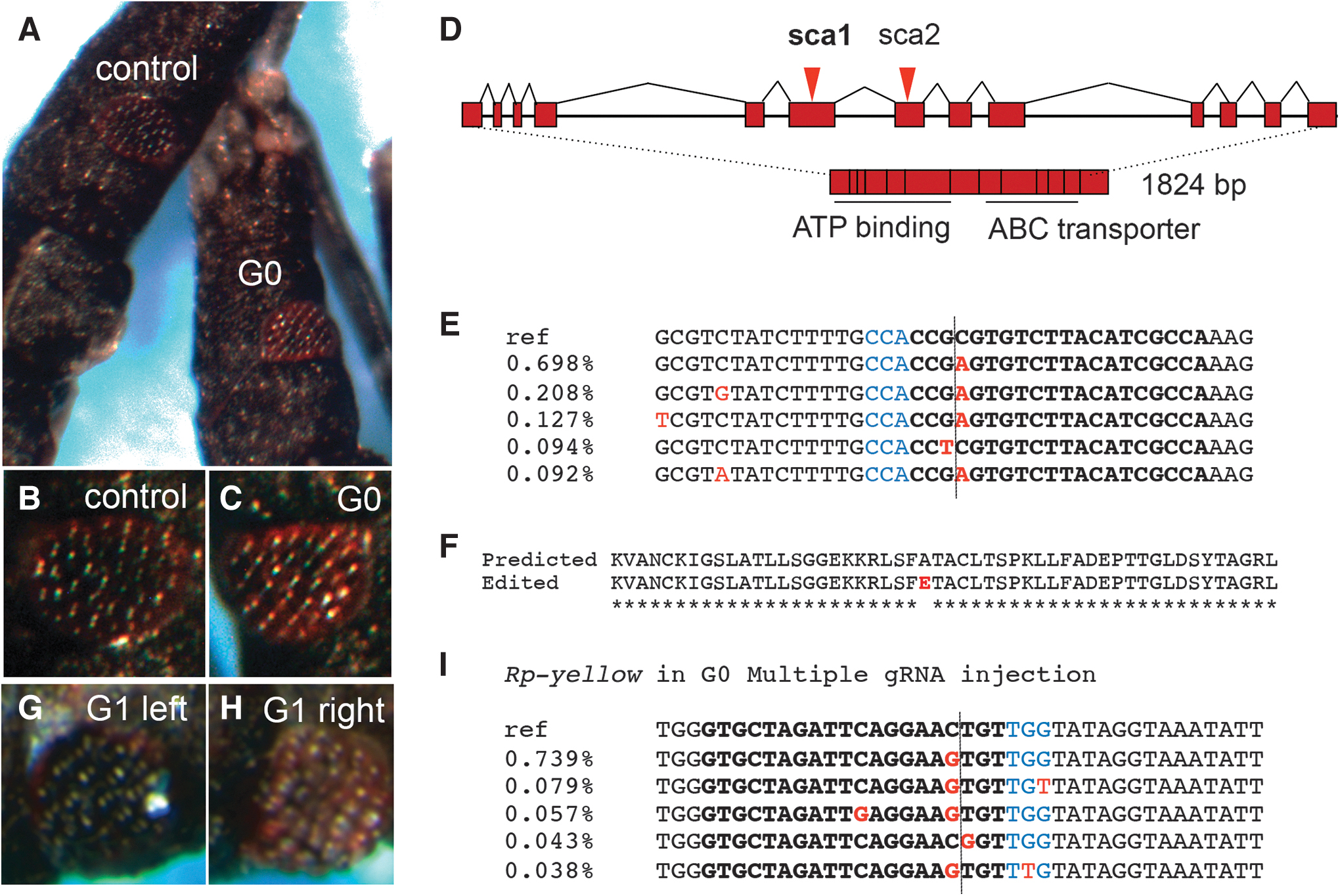
Total genomic DNA from one G0 third-instar nymph displaying an entirely yellow cuticle (Fig. 2) was extracted and sequences surrounding the Rp-y gRNA target site were PCR amplified and cloned into a TOPO TA cloning vector (Invitrogen) for bacterial transformation. Single colonies were Sanger sequenced with M13 or specific primers from both sides of the target site. We detected a large deletion in 3/10 colonies sequenced. This lesion is predicted to generate a truncated and nonfunctional protein (Fig. 2D, E). Importantly, we observed an 11 bp deletion located 24 bp 3′ of the Cas9 cleavage site in 3/8 control animals and 13/30 injected animals, characterizing heterozygotes for a deletion that is present in the colony. This 11 bp deletion resides inside an intron, is not predicted to alter protein function, and does not result in a visible phenotype, since animals homozygous for the deletion display no phenotype. Despite apparently not altering gene function, the deletion in heterozygotes precludes the use of heteroduplex assays for the screening of nearby lesions generated by CRISPR.

CRISPR-directed gene edition events in the yellow locus.
The large deletion surrounding the Cas9 cleavage site in the yellow nymph (y29) is surprising, since NHEJ events expected to correct DSBs frequently generate small indels. However, large deletions resulting from CRISPR-Cas9 gene editing have been reported in ticks. 18 To determine the different types of lesions resulting from the CRISPR editing process, we amplified and performed next-generation deep sequencing of the region surrounding the gRNA target site. Small indels were observed at low frequencies, as well as a series of larger deletions (Fig. 2F and Supplementary Table S4). These deletions are consistent with that observed by Sanger sequencing and indicate that y29 is a mosaic containing a mixture of wild-type Rp-yellow alleles and alleles that harbor deletions and base changes surrounding the target site, that likely impair Rp-yellow function.
Analysis of the sequences surrounding the Rp-white gRNA target site in the white-eyed nymph, on the other hand, displayed no large sequence modifications (Fig. 3A–D). Sanger sequencing of the nymph genomic DNA revealed no modification relative to controls, but a small indel was detected by PCR amplification and cloning in 1/10 colonies. However, the C > A base change is located 71 bp 3′ of the predicted Cas9 cleavage site and may represent a rare SNP. Deep sequencing revealed a small fraction of sequences displaying single nucleotide modifications at the Cas9 cleavage site (Fig. 3D and Supplementary Table S5). The most frequent of these sequences was predicted to encode an amino acid substitution, replacing a tryptophan with a cysteine residue, a significant alteration that could potentially impair protein function (Fig. 3E).

Gene edition in the white locus leads to loss of eye color.
Subsequently, we examined nymphs for phenotypes likely resulting from impairment of Rp-sca function. Four nymphs displayed a red eye phenotype, one with only slightly red eyes (from P2C-Cas9 injection and were not analyzed further) and three individuals with strong red eye phenotypes (from independent BtKV-Cas9 injected mothers). One of these three individuals was derived from a single Rp-sca gRNA injection and the other two from injections with gRNAs against multiple genes (Fig. 4A–C). We chose one of the multiple gRNA-injected nymphs for Illumina deep sequencing. Analysis of the sequences surrounding the Rp-sca gRNA target site revealed a significant amount (3.3% of aligned reads) of base substitutions located three to four nucleotides from the PAM site, indicating likely imprecise repair through the NHEJ pathway (Fig. 4E). The most frequently observed nucleotide substitution is predicted to result in an amino acid replacement from an alanine to a negatively charged glutamic acid residue (Fig. 4F). Since the gRNA target site is placed in the region that encodes the ATP binding site, this substitution could impair protein function leading to the impaired transport of dark pigments in the eye. 19 Although none of the red-eyed G0 nymphs survived to adulthood, one G1 animal resulting from crosses among the brothers and sisters of the red-eyed G0 (in Fig. 4A, C) displayed a mosaic eye color phenotype, wherein one eye was red, while other eye had ommatidia as black as the wild type (Fig. 4G, H). We did not detect edited alleles by Sanger sequencing, suggesting that the animal might be a mosaic with a small number of edited alleles, arguing against the hypothesis that it resulted from gene editing events transmitted through the germ line.
Red eyes result from scarlet gRNA/BtKV-Cas9 single and multiple gRNA injections. First, instar siblings resulting from a female injected with multiple gRNAs (directed to Rp-sca, Rp-y, Rp-w, and Rp-aaNAT) in BtKV-Cas9 RNPs, showing a wild type
Next, we investigated whether a visible phenotype resulting from genome editing in one locus (Rp-sca) could help identify coediting events in other loci that would not generate any visible effect. Therefore, we sequenced the Rp-w and Rp-y gRNA target sites in the red-eye nymph resulting from the multiple gRNA ReMOT. Base substitutions were detected along a guanine stretch in the Rp-w target site, three nucleotides from the PAM, both in the G0 and the G1 nymph. However, a similar percentage of modified sequences were observed in a control nymph that was sequenced under identical conditions, suggesting that these variant alleles did not result from Cas9-mediated editing events (Supplementary Fig. S3). On the other hand, sequence analysis for Rp-y in G0 identified several altered sequences (1.84% of reads, as compared to the control 0.41% modified sequences) at the predicted Cas9 cleavage site (Fig. 4I and Supplementary Fig. S3), despite the lack of a detectable cuticle phenotype expected to result from a putative loss of Rp-yellow function. In addition, G1 Rp-y sequencing identified a series of deletions surrounding the gRNA target site (2.58%) that was represented in yet a much smaller percentage in control wild-type sequences (0.06%) (Supplementary Fig. S3).
We note that the multiple gRNA-injected nymphs analyzed were first instars that were not allowed to develop to further stages. Thus, we could not confirm whether a visible cuticle phenotype resulting from these alterations in the Rp-y locus would appear at later stages, similar to what we observed with the y29 third instar nymph shown in Figure 2.
Discussion
ReMOT control is more effective than DIPA-CRISPR in delivering CRISPR cargo to triatomine oocytes
CRISPR-Cas9 mutagenesis has recently emerged as an efficient method for genome engineering in a number of arthropod species. Most methodologies involving CRISPR rely on the embryonic delivery of CRISPR elements by microinjection. However, in animals that lay eggs encased in a hard chorion such as triatomines, or other types of structures that preclude embryonic delivery of CRISPR elements such as the cockroach ootheca, maternal delivery may be a more accessible method for gene edition. Here, we tested oocyte delivery of RNP CRISPR complexes containing untagged and tagged Cas9 protein injected into the female hemocoel. First, we showed that untagged mCherry protein was able to enter vitellogenic oocytes, but that fusing mCherry to ovary targeting sequences delivered significantly more protein.
Using the DIPA-CRISPR method (untagged Cas9), we did not detect any animals displaying cuticle or eye color phenotypes resulting from injections with untagged Cas9 (7 females injected, 236 nymphs screened), while with the ReMOT control (using Cas9 fused to two different ovary targeting peptides), we detected multiple editing events in offspring. Optimization of either method may increase the efficiency of genome editing, but, currently, our results indicate that ReMOT control is a more effective method than DIPA-CRISPR for genome editing in R. prolixus. The greater amount of oocyte fluorescence resulting from hemocoel injection of BtKV-mCherry relative to untagged mCherry in T. infestans suggests that this greater efficiency may hold true for other triatomine vectors of Chagas disease.
Visible markers for genome edition in R. prolixus
Pigmentation mutants have been classically used as genetic markers in recombination experiments and genetic crosses since the early days of Drosophila genetics. 25 Pigmentation genes are also frequently carried as visible markers in transgenes, such as a forshortened mini-white version of the w gene in Drosophila 26 and kynurenine monooxygenase gene in Aedes. 27 Cuticle and eye color loci may also be very useful in co-CRISPR experiments to generate alleles in loci that may not engender visible phenotypes on their own.28,29 In such cases, individuals exhibiting pigmentation phenotypes may have increased chances of displaying lesions in a second locus that is targeted in parallel. Here, we have shown that visible phenotypes can be generated by CRISPR-mediated genome editing of pigmentation loci in R. prolixus, corroborating previous evidence from gene knockdown studies. 19 Three loci (Rp-y, Rp-w, and Rp-sca) display easily scored phenotypes in the eye and cuticle resulting from CRISPR lesions. Visible phenotypes are seen since the first instar for Rp-w and Rp-sca but may be harder to detect in young Rp-y mosaics. For instance, the light color of the y29 animal was only perceived at third larval instar stage, possibly due to maternal provision of Rp-y messages obscuring the mutant phenotype in younger first instars. Notwithstanding, the use of a cuticle marker may be best suited to accompany some types of loss-of-function experiments, such as behavioral experiments that require visible cues. Among the four loci tested, no phenotype for Rp-aaNAT was observed, despite the striking dark phenotype resulting from knockdown experiments with RNA interference. 19 This may be due to inefficiency of the gRNA, lethality resulting from gene knockout, or to other limitations.
As mentioned above, ReMOT control delivery of multiple gRNAs indicates that the parallel delivery of marker gRNAs such as that for Rp-sca may help identify gene edition events in loci that are not expected to generate a visible phenotype. This procedure, termed co-CRISPR, has been shown to be effective in C. elegans, based on a worm movement phenotype, 30 and in Drosophila, where ebony 31 and ovoD 28 enhanced the screening and identification of genome-editing events. Among the loci herein tested, Rp-sca was most effective in generating animals with a visible phenotype. Importantly, Rp-sca knockdown does not alter viability, 19 indicating that knockouts may be viable as well. Conversely, Rp-w KD decreases viability in R. prolixus and Of-w loss of function is lethal in the Hemiptera Oncopeltus faciatus. 32 These differences in viability may be irrelevant while screening G0 mosaics but could lead to loss of edited G1 individuals if Rp-w were used.
Gene editing efficiency in the kissing bug parallels that reported for other insects
Two important characteristics define the suitability of gene editing methods for mutagenesis: the efficiency, defined by the number of edited animals in a population, and germ line transmission of induced mutations. Due to the long generation time for R. prolixus (∼6 months) and the limited number of gene-edited individuals capable of surviving this protracted period, determining germ line transmission is extremely challenging. Therefore, we defined editing efficiency at G0, taking into account that animals displaying visible phenotypes are likely mosaics carrying biallelic lesions at target loci.
Considering the different target loci, we find an editing efficiency (number of edited animals in the population) of 0.7–4.0% in the G0s, resulting from ReMOT control with BtKV-Cas9. This is within the range reported for ReMOT delivery in Aedes aegypti (1–2.5% 14 ), Anopheles stephensi (∼2%, 15 ), Ixodes scapularis (1.7–4.2%, 18 ), Culex pipiens (∼2%, 33 ), and B. tabaci (0.2–2% 16 ), as scored in the G0. It is important to take into account that the editing efficiencies reported for the species above were defined solely based on visible phenotypes. Conforming to this practice, screening total genomic nymph DNA by Sanger sequencing has proven unsuitable to identify gene-editing events in R. prolixus, as it revealed no detectable sequence modifications, even in mosaic animals displaying visible phenotypes. This indicates that the proportion of edited cells in animals displaying visible phenotypes is likely to be small in relation to wild-type cells of the entire animal, consistent with the modest percentage of edited sequences revealed by deep sequencing. Since similar G0 editing efficiencies reported are correlated with germ line transmission of gene editing events in several species listed above, future experiments with large numbers of injected R. prolixus females may enable recovery of heritable germ line-transmitted edited alleles in this species.
Due to the long R. prolixus life cycle and the limited number of edited animals obtained, we were yet not able to identify germ line transmission of edited events. The unexpected identification of a G1 nymph with one sole red eye is suggestive of some type of partial loss of function allele transmitted through the germ line.
However, Sanger sequencing for the entire nymph or for PCR-amplified sequences cloned into bacterial vectors revealed no detectable sequence modifications, indicating that the visible phenotype results from a small number of somatic editing events. If correct, this could imply that Cas9/gRNA complexes can be transmitted from one generation to another, from the BtKV-Cas9 injected mothers (G-1), through G0, to G1. Although surprising, particularly considering the long R. prolixus generation time, a similar form of “shadow drive,” wherein Cas9/gRNA complexes are transmitted to progeny in the absence of gRNA or Cas9 transgenes, has been reported in Drosophila. 29 Furthermore, we have observed transmission of an expression of DNA plasmid for two generations post-ReMOT injection in An. stephensi (Macias and Rasgon, unpublished). As such enduring effects would have strong implications for biological vector control, future studies should be undertaken to investigate the possibility of transgenerational delivery of gRNA/Cas9 complexes in triatomines.
Challenges for genome edition in R. prolixus
In addition to their long generation time, we found other challenges for gene editing in R. prolixus. One quite unexpected observation was sequence heterogeneity. Despite the long inbreeding of our R. prolixus colony, we found multiple deletions carried as heterozygotes in target loci. As discussed above, this characteristic impairs the use of heteroduplex analysis to screen for editing events. Unfortunately, isogenic lines are too weak for use in these types of experiments, indicating that one should sequence many insects before designing gRNAs for gene editing and that sequencing should not be restricted to the gRNA target region, but should also cover any nearby region that will be PCR amplified during screening.
Another challenge will be to define conditions for repair by HDR, to enable the precise modification of target sequences and to incorporate exogenous DNA. Currently, ReMOT control only enables gene edition and repair by NHEJ. This limitation could be solved in the future through the development of effective embryo delivery of CRISPR elements or alternative methods including maternal delivery of RNPs associated with exogenous sequences containing homology arms to sequences flanking the Cas9 cleavage site.
Footnotes
Acknowledgments
We would like to thank Dr. Pedro L. Oliveira for helpful comments on the manuscript. We are grateful to the animal facility at the Institute of Medical Biochemistry for technical assistance with R. prolixus husbandry and to the International Reference Laboratory for Triatomine taxonomy for providing T. infestans animals.
Authors' Contributions
L.L., M.B., J.M., and R.C. collected the data. D.B., A.L., H.A., and Z.L. performed data analysis and interpretation. V.M., J.L.R., and E.B. contributed analysis tools. H.A. and V.M. conceived the original idea. H.A. and M.B. designed the analysis. H.A. and E.B. supervised the project. H.A. drafted the first version of the manuscript. All authors revised and approved the final version of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, 88881.117632/2016-01; https://www.gov.br/capes/pt-br) to H.A. and E.B., and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ, E-216/10.101034/2018; https://www.faperj.br) to H.A. H.A. is a CNE FAPERJ researcher and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; https://www.gov.br/cnpq/pt-br) fellow. L.L. and J.M. were supported by graduate fellowships from CAPES, R.C. was supported by an undergraduate fellowship from CNPq and M.B. and D.B. were supported by postgraduate fellowships from FAPERJ. J.L.R. was supported by NSF (https://www.nsf.gov) grant 1645331, USDA (![]() ) Hatch funds (Project No. 4769), and funds from the Dorothy Foehr Huck and J. Lloyd Huck endowment.
) Hatch funds (Project No. 4769), and funds from the Dorothy Foehr Huck and J. Lloyd Huck endowment.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.