
Abstract
Antibiotic resistance poses a global health crisis limiting the efficacy of available therapeutic agents. We explored CRISPR-Cas-based antimicrobials to combat multidrug resistance in methicillin-resistant Staphylococcus aureus (MRSA), targeting methicillin (mecA), gentamicin (aacA), and ciprofloxacin (grlA, grlB) resistance genes. Engineered CRISPR plasmids with specific single-guide RNAs were electroporated into MRSA strains. Real-time polymerase chain reaction assessed gene expression changes, while antibiotic susceptibility tests (ASTs) evaluated resistance status. Results showed a 1.5-fold decrease in mecA, a 5.5-fold decrease in grlA, a 6-fold decrease in grlB, and a 4-fold decrease in aacA expression. ASTs demonstrated the reversal of resistance to beta-lactam, quinolone, and aminoglycoside antibiotics. Western blot analysis revealed a 70% decrease in penicillin-binding protein 2a expression. Sanger sequencing confirmed point mutations in the grlB and aacA genes. Our findings highlight the potential of CRISPR-Cas9 technology to restore antibiotic efficacy against multidrug-resistant pathogens.
Introduction
Antibiotic resistance is a significant global challenge, ranking among the top 10 threats identified by the World Health Organization (WHO). It poses not only a serious health crisis but also a socio-economic burden due to the high rates of mortality and morbidity associated with resistant infections.1–3 In 2019, antibiotic resistance was linked to approximately 5 million deaths worldwide, highlighting the urgent need for effective interventions.3,4 Projections indicate that by 2050, this issue could lead to an annual death toll of 10 million people and a 3.8% reduction in global gross domestic product, potentially affecting 24 million individuals. 5
Despite ongoing efforts to develop new antibiotics, the pharmaceutical industry faces substantial hurdles, including the lengthy and expensive process of drug development and the rapid emergence of resistance to new antimicrobial agents. To address these challenges, researchers are exploring alternative strategies such as drug repositioning and targeting bacterial metabolic or virulence pathways.6–8 However, existing antibiotics often lack the specificity required to selectively eliminate pathogenic bacteria, leading to disruptions in beneficial host microbiota.9–12 This underscores the global need for more precise, targeted therapeutic approaches. 13
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology, paired with CRISPR-associated (Cas) proteins, has transformed genetic engineering since its discovery as an adaptive immune system in bacteria and archaea. Since 2012, the CRISPR-Cas9 system has become widely adopted due to its simplicity, precision, and versatility in genome editing.14–16 Cas9, an RNA-guided endonuclease, introduces double-strand breaks at specific genomic locations, enabling targeted gene disruption, insertion, or correction.16,17 The ability to design multiple single-guide RNAs (sgRNAs) to simultaneously target different genes makes CRISPR-Cas9 an especially powerful tool for multiplex gene editing. 18 These advances in genetic engineering present a promising new avenue for developing targeted antimicrobials against resistant microorganisms.
Our study, driven by the escalating global health crisis of antibiotic resistance and diminishing therapeutic options, focuses on the potential of CRISPR-Cas9 technology to address this issue. Specifically, we target methicillin-resistant Staphylococcus aureus (MRSA), a pathogen identified by the WHO as critically requiring new antibiotic development and research. 19 MRSA’s genetic adaptability and virulence factors make it a serious threat, capable of causing a wide range of infections, from mild to life-threatening. 20 Penicillin-binding proteins (PBPs) are essential for the last steps of the synthesis of peptidoglycan in the bacterial cell wall. Staphylococcus aureus encodes four different PBPs, named PBP1–4, which can be targeted by β‐lactams. MRSA strains encode, in addition to PBP1–4, a fifth PBP protein known as PBP2a, which is responsible for the resistant phenotype. 21 The core of methicillin resistance in MRSA lies in the PBP2a protein, encoded by the mecA gene on the staphylococcal cassette chromosome mec, absent in susceptible strains, which renders most MRSA strains resistant to a broad spectrum of antibiotics.22,23
Our research targets the mecA gene responsible for methicillin resistance, the aacA gene linked to gentamicin resistance, and the grlA and grlB genes associated with ciprofloxacin resistance in clinical MRSA isolates. We designed multiple sgRNAs to target these resistance genes and cloned them into the pCasSA vector using Golden Gate cloning, which allows the insertion of multiple sgRNA sequences simultaneously. The pCasSA vector was engineered to express all four sgRNAs under separate promoters, ensuring efficient targeting of each gene. Cas9 was selected for this study due to its extensive validation across various organisms and cell types, including bacteria, and its robust targeting efficiency and versatility. 16 Unlike Cpf1, Cas9’s NGG PAM sequence is more commonly found in bacterial genomes, simplifying target site selection. The ability to design multiple sgRNAs for concurrent gene targeting makes Cas9 particularly well-suited for our goal of disrupting multiple antibiotic resistance genes. 18 The engineered plasmid was introduced into MRSA cells, and successful transformants were selected using antibiotic resistance markers. We validated the effectiveness of multiplex gene editing by sequencing the target regions of all four genes, confirming the presence of indels and mutations. In addition, quantitative PCR and western blot analyses were performed to assess reductions in gene expression and protein levels. Our overarching goal is to sensitize these MRSA isolates to antibiotics that have lost efficacy due to resistance, providing a promising strategy for tackling multidrug-resistant pathogens.
Materials and Methods
Bacterial strains, plasmid, and primers
Twenty S. aureus strains isolated from clinical samples at the Department of Medical Microbiology of Ege University Hospital were included in this study. Staphylococcus aureus ATCC 29213 was used as the control strain. All bacterial isolates were stored in a Brain-Heart Infusion broth medium with 10% glycerin at −80°C until the study. pCasSA was a gift from Quanjiang Ji (Addgene plasmid # 98211). The primers used in the study are listed in Supplementary Table S1.
Identification of isolates and determination of antibiotic susceptibilities
Identification and antibiotic susceptibilities of clinical isolates were performed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (Biomerieux, France) and VITEK-2 Compact (Biomerieux, France) automated system, respectively. VITEK-2 test results were confirmed by broth microdilution testing. mecA, grlA, grlB, and aacA resistance genes in clinical isolates resistant to methicillin, ciprofloxacin, and gentamicin were detected by conventional polymerase chain reaction (PCR). The details of the clinical isolates are provided in Supplementary Table S2. Among these isolates, one representative clinical MRSA isolate was selected that contained all of the target antibiotic resistance genes (mecA, grlA, grlB, and aacA) and was found to be resistant to these antibiotic groups and was identified to be in a different group by clonal relatedness (Strain ID: 1). A representative clinical MRSA isolate was selected and used in the study. CRISPR editing was performed in this isolate.
Vector construction
The CRISPR-Cas9 system used in this study is based on the pCasSA vector, which contains the Cas9 nuclease and the necessary components for sgRNA expression. pCasSA (plasmid 98211; Addgene) was a gift from Q. Ji. The vector was engineered to include multiple sgRNA sequences targeting different resistance genes (mecA, aacA, grlA, and grlB).
sgRNA binding locations
Specific sgRNAs were designed to target conserved regions within the mecA, aacA, grlA, and grlB genes. DNA sequences of all guide RNAs (gRNAs) used for gene knockout are listed as 5′–3′ sequences in Supplementary Table S3. All sequences were selected to precede the 5′-NGG protospacer-adjacent motif sequence. The target sequences for each sgRNA were chosen based on high on-target scores and minimal off-target effects, which were verified using the Benchling software. On-target activity score and off-target specificity scores were calculated according to Doench et al. (2014) and Hsu et al. (2013), respectively.24,25 On-target and off-target scores were from 0 to 100. Higher is better. The on-target and off-target values of the sgRNAs used in this study are shown in Supplementary Table S4. Each sgRNA was cloned into the vector using Golden Gate assembly, which allows for the simultaneous insertion of multiple sgRNA sequences into distinct expression cassettes under the control of separate promoters.
mecA Gene: sgRNA targeting mecA was designed to bind at positions 432–452 bp of the coding sequence.
aacA Gene: sgRNA targeting aacA was designed to bind at positions 50–70 bp of the coding sequence.
grlA Gene: sgRNA targeting grlA was designed to bind at positions 87–107 bp of the coding sequence.
grlB Gene: sgRNA targeting grlB was designed to bind at positions 84–104 bp of the coding sequence.
These binding locations were selected based on their proximity to conserved regions, ensuring efficient targeting with high on-target activity and minimal off-target effects, as confirmed through Benchling software analysis.
A schematic diagram of the pCasSA vector showing the Cas9 gene, sgRNA cassette, antibiotic resistance markers, and origin of replication was included in Figure 1a. An experimental set-up schematic consisting of performed studies is shown in Figure 1b.
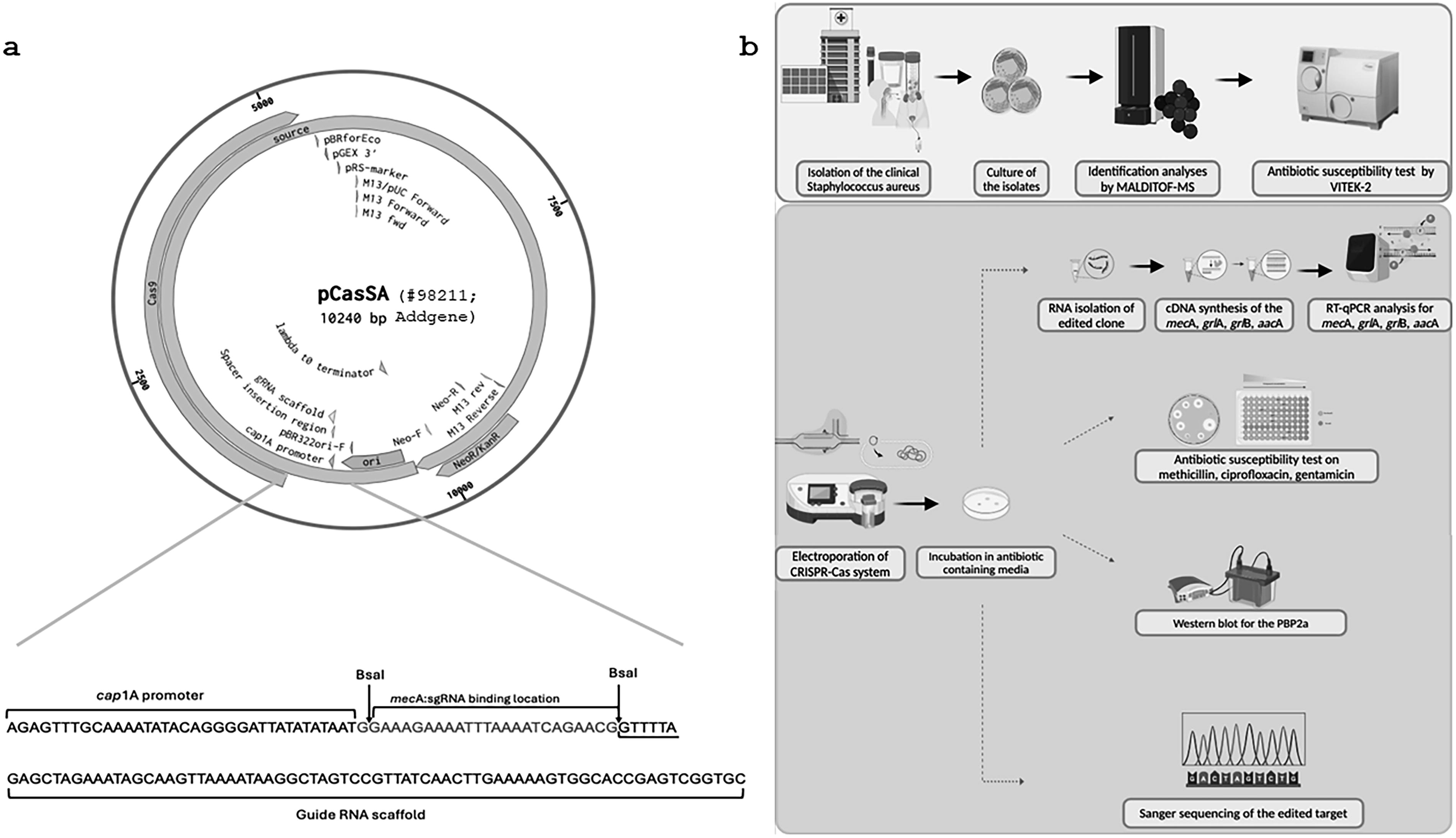
Restricted digestion
To reduce the risk of false positive results in the ligation of gRNAs into the plasmid, the plasmid was linearized with BsaI (NEB, UK). The linearized product was visualized by electrophoresis in 1% agarose gels and then purified from agarose gel. Purification was performed using the Plus Gel Eluted Kit (GMbiolab, Taiwan).
Phosphorylation, annealing of oligos, and plasmid ligation
On-target and off-target scores were considered, and oligos specific to each target site were designed from Benchling.com. These oligos were synthesized with GAAA insertion at the 5′ end of the first sequence and CAAA extension at the 5′ end of the second chain to bind to the sticky ends formed in the plasmid after restriction digestion (Supplementary Table S3). To reduce the possibility of false positives, oligos were inserted by ligation into a linearized plasmid. The ligation product was transferred into Escherichia coli DH10B chemically competent cells by heat shock transformation. After transformation, cells were inoculated on Luria-Bertani (LB) agar containing 50 μg/mL kanamycin. After incubation at 37°C, sample colonies were selected on LB agar containing 50 μg/mL kanamycin for plasmid isolation. Isolation was performed using Monarch Plasmid Miniprep Kit (NEB, UK).
Colony PCR
After plasmid isolation, colony PCR was used to confirm whether the targeted ligation occurred in the isolated plasmids. For this purpose, colony PCR was performed to amplify a 120-bp region with appropriate primers, with the forward primer being the site of plasmid ligation.
Preparation of electrocompetent MRSA cells
A single colony of MRSA was inoculated into Tryptic Soy broth (TSB) medium. After 18 h of incubation, an appropriate volume of the broth culture was taken and diluted 1/100 in TSB medium and incubated in a shaking incubator at 30°C for 2–3 h (i.e., OD600: ∼0.4). When the targeted OD600 value was reached, the culture was incubated in dry ice for 10 min. The culture was centrifuged at 5000 rpm for 5 min. The supernatant was removed, and cells were suspended in a 500 mM sucrose solution. After centrifugation and re-suspension steps were repeated twice, the supernatant was discarded and the pellets were suspended in 0.5 M sucrose, aliquoted, and stored at −80°C until electroporation. 26
Electroporation
The electrocompetent MRSA suspension was mixed with pCasSA (∼2 μg) containing the insert unit appropriate for the target genes by gently tapping the tube. The mixture was transferred to a 1 mm electroporation cuvette at room temperature. The cells in the cuvette were pulsed using Eporator (EPPENDORF—Eporator, Germany) at 21 kV/cm, 5.0 ms. After electroporation, the cells were incubated in a TSB medium at 30°C for 1.5 h. The transformant cell culture was then inoculated with a Drigalski spatula into a Tryptic Soy agar (TSA) medium containing 5 μg/mL chloramphenicol. The plates were incubated at 30°C for 48 h. 26
Investigation of expression levels of resistance genes by quantitative real-time reverse transcriptase PCR
RNA isolation from wild-type (WT) clinical MRSA isolate and knock-out (KO) strain was performed using the Total RNA purification kit (GMbiolab, Taiwan). Concentrations and purities of RNA isolates were measured on a Beckman Coulter DU-730 (USA). The cDNA was synthesized from RNA at the appropriate concentration using the Onescript Plus Reverse Transcriptase cDNA Synthesis Kit (ABM, USA) according to the manufacturer’s recommendations. The cDNAs synthesized from total RNAs were used for real-time reverse transcriptase quantitative PCR (RT-qPCR) with a 2× Magic SYBR Mix (Procomcure, Australia) kit. cDNA concentrations were measured, equalized, and then added to a 96-well Armadillo (Thermo Scientific, USA) microplate. The master mix was added, and the plate was placed in a LightCycler 480 II (Roche). The results were evaluated by the ΔΔCT method considering the Cp values obtained from LightCycler 480 Software release 1.5.0. SP3. The primers for mecA, aacA, grlA, and grlB, and housekeeping gene 16srRNA utilized in qPCR experiments are shown in Supplementary Table S1. Using 16S rRNA as a housekeeping gene, changes in the expression levels of antibiotic resistance genes were statistically evaluated by normalization with the ΔΔCT method.
Phenotypic ASTs
The effect of genomic modification by CRISPR-Cas technology on the resistance profile was examined by phenotypic ASTs. Broth microdilution and disk diffusion methods for WT and KO isolates were performed according to the European Committee on Antibiotic Susceptibility Testing (EUCAST) guidelines. Staphylococcus aureus ATCC 29213 was used as the control strain.
Broth microdilution method
The broth microdilution method was performed following EUCAST and ISO (20776) criteria. The turbidities of WT and KO strains were adjusted to McFarland 0.5 with 0.9% saline. Bacterial inoculums were diluted 1/100 with MHB to a final concentration of 5 × 105 CFU/mL. Ciprofloxacin, oxacillin, and gentamicin solutions with final concentrations (2048 μg/mL) were prepared with sterile distilled water. MHB was added to all wells of a U-bottom 96-well sterile microplate, and then oxacillin, gentamicin, and ciprofloxacin solutions were added only to the first wells of the microplate. Serial dilution was performed. Bacterial suspensions were inoculated into the wells. Sterility control and growth controls were included. After incubated at 37°C for 24–48 h, the lowest concentration without growth was determined as the minimum inhibitory concentration (MIC-μg/mL).27,28
Disk diffusion method
WT and KO strains were adjusted to standard turbidity of McFarland 0.5 with 0.9% saline. The strains were inoculated onto an Mueller-Hinton Agar (MHA) medium with a swab. After the media were allowed to dry for a few minutes, cefoxitin (30 µg-FOX-30), gentamicin (10 µg-CN-10), and norfloxacin (10 µg-NOR-10) disks were placed on the media using sterile forceps. After incubation at 37°C for 24–48 h, the zone diameters around the disks were measured and evaluated. 29
Protein purification and western blot
Western blotting was performed to evaluate the expression change in PBP2a protein in KO strains. For this purpose, protein isolation was performed from WT and KO strains. 30 The Bradford Commasie Blue method was used to determine protein concentrations. Pierce™ Coomassie (Bradford) Protein Assay Kit (Thermo Scientific) was applied according to the manufacturer’s recommendations. Protein concentrations were calculated using absorbance values. Proteins were brought to equal concentrations before loading (11 μg). A protein molecular weight marker (GenemarkBio, Taiwan) was added to the first well, and WT protein and KO protein mixture were added to the other wells in triplicate. Electrophoresis was performed with a Bio-Rad Protean-II electrophoresis system at 80 V for 10 min, then 100 V for 120 min. After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was completed, proteins were transferred to polyvinylidene difluoride (PVDF) (Thermo Scientific) membranes (+4°C, 100 V, 2 h). Nonspecific binding was prevented after transfer. Then, the primary antibody (mouse anti-PBP2a, Raybiotech, USA) was diluted 1/1000 in 5% milk powder and added to the membrane. The membrane was then incubated overnight at +4°C on an orbital shaker. At the end of the incubation, the membrane was washed three times with 1XTBS-T for 10 min incubations. A secondary antibody (anti-mouse IgG, Jackson ImmunoResearch, UK) diluted 1/10,000 was added to the membrane and incubated on a shaker at room temperature for 2 h. At the end of incubation, the membrane was washed three times with 1XTBS-T at 10-min intervals. Luminol/Enhancer from the chemiluminescence substrate kit (SuperSignal West Pico Plus, Thermo Scientific) and Peroxidase Buffer were added to the membrane in a 1:1 ratio and kept on the shaker for 5 min. Imaging was performed with Fusion FX-7 (Vilbert Lourmat, France).
Sanger sequence analysis
Sanger sequencing of the WT strain was conducted for the mecA, aacA, grlA, and grlB gene regions to confirm that no pre-existing mutations were present. These sequences were compared with the NCBI database (Accession numbers: BA000017, AP003367.1, and D67075.1) to verify consistency and ensure that the observed mutations in the KO strain were exclusively the result of CRISPR-Cas9 editing. Genomic DNA isolation of KO S. aureus strains was performed using a Nucleospin Microbial DNA kit (Macherey-Nagel, Germany). To examine the mutation caused by the CRISPR-Cas system in the target gene regions, regions of approximately 1000 bp were selected to include each gene region. The relevant regions were amplified by PCR using primers designed for this study and shown in Supplementary Table S1 Q5 High Fidelity DNA Polymerase (NEB) was used for sequence analysis with appropriate purity and accuracy.
Statistical analysis
All experiments were conducted in triplicate. Standard deviations (SDs) are calculated by GraphPad Prism 5 software (San Diego, CA, USA). Statistical calculations were analyzed by one-way ANOVA tests. p < 0.5 was accepted as statistically significant.
Results
Quantitative RT-qPCR
Multiple transformants were obtained, and each was subjected to AST, RT-qPCR, and Sanger sequencing. All transformants consistently exhibited the expected phenotypic changes, such as increased antibiotic susceptibility and reduced expression of targeted resistance genes. Sanger sequencing revealed similar point mutations and small indels across all transformants, indicating consistent CRISPR-Cas9-mediated disruption of the target genes.
We aimed to assess how antibiotic resistance would change through the multiplex knockout of a representative MRSA clone containing four antibiotic resistance genes (mecA, grlA, grlB, and aacA). To quantitatively evaluate changes in gene expression, we utilized RT-qPCR with 16S rRNA as the housekeeping gene. Expression levels of the targeted genes were statistically analyzed using the ΔΔCT method, which provided a reliable framework for comparisons. Figure 2 shows the gene expression changes in KO strains compared to WT strains. In the KO strain, we observed a significant impact on the expression of key resistance genes. Relative to the WT strain, mecA gene expression decreased by 1.5-fold, reflecting the successful targeting and suppression of methicillin resistance. In addition, the KO strain displayed a 5.5-fold reduction in grlA gene expression, a 6-fold reduction in grlB gene expression, and a 4-fold reduction in aacA gene expression compared with the WT strain. These findings emphasize the precision and effectiveness of CRISPR-Cas9-mediated genome editing in selectively modulating the expression of resistance genes. The observed decreases in gene expression indicate a successful alteration in the resistance profile of the KO strains, highlighting the potential of CRISPR technology to combat antibiotic resistance.

Changes in the expression ratio of related genes in RT-PCR, *p < 0.01, ***p < 0.001. KO, knock-out; RT-PCR, real-time polymerase chain reaction; WT.
Phenotypic ASTs: Broth microdilution and disk diffusion method
The results of broth microdilution and disk diffusion tests for the clinical and control strains were evaluated in Tables 1 and 2, following EUCAST guidelines. The phenotypic behavior of the KO strain was examined using both broth microdilution and disk diffusion methods. Antibiotic susceptibility profiles of the KO strain were compared with the corresponding WT strain. The KO strain exhibited a significant reduction in MIC values for oxacillin, gentamicin, and ciprofloxacin, confirming the successful disruption of the targeted resistance genes (Table 1). ASTs revealed a dramatic transformation in the resistance profile of the WT strain, which was resistant to all antibiotics tested. In contrast, the CRISPR-Cas9-engineered KO strain demonstrated a clear shift toward antibiotic susceptibility. Notably, the cefoxitin zone diameter increased substantially, accompanied by a 128-fold reduction in oxacillin MIC values, signifying the elimination of methicillin resistance. CRISPR-Cas9’s precision in disrupting resistance mechanisms was further validated by a significant reduction in ciprofloxacin MIC values and an increased norfloxacin zone diameter, leading to the reclassification of the strain as susceptible, effectively abolishing ciprofloxacin resistance. In addition, aminoglycoside resistance was decisively broken, as evidenced by a notable increase in the gentamicin zone diameter and a 512-fold reduction in MIC values (Tables 1 and 2). These phenotypic changes were directly attributed to the targeted suppression of the mecA, grlA, grlB, and aacA genes, resulting in the cessation of resistance to methicillin, quinolones, and aminoglycosides. These results conclusively demonstrate the efficacy of CRISPR-Cas9-mediated genetic intervention in converting antibiotic resistance to susceptibility. The successful elimination of multiple drug resistances underscores the potential of this innovative approach in addressing the formidable challenge of antibiotic resistance.
Minimum inhibitory concentration in broth microdilution
MRSA, methicillin-resistant Staphylococcus aureus.
Diameter of inhibition zone in disc diffusion
Western blotting
The CRISPR modification targeting the mecA gene resulted in significant effects on both gene expression and phenotypic resistance, as demonstrated by a comprehensive series of analyses. RT-PCR revealed a substantial reduction in mecA gene expression, confirming the successful modulation of methicillin resistance at the molecular level. Subsequent phenotypic ASTs further validated these findings, demonstrating the complete abolition of methicillin resistance in the KO strains. To investigate the impact of the mecA gene modification further, western blot analysis was performed to assess the expression of PBP2a. Figure 3 highlights a marked decrease in PBP2a expression in the KO-mecA strain, underscoring the effectiveness of the CRISPR-mediated genetic intervention. Image analysis using ImageJ 8 software, with normalization based on Spa, provided precise quantification of the observed expression changes. As shown in Figure 4, the KO strain exhibited a striking 70% reduction in PBP2a expression compared with the WT strain, reinforcing the successful disruption of methicillin resistance. The reduction in PBP2a levels is directly attributable to mutations induced in the mecA gene region by CRISPR technology. These results not only demonstrate the efficacy of CRISPR technology in achieving targeted genetic modifications but also highlight its transformative potential in combating antibiotic resistance at both the genetic and phenotypic levels. The ability to modulate the expression of key resistance genes offers promising prospects for developing precision therapies against multidrug-resistant pathogens.

Western blot membrane image (KO-mecA, mecA gene suppressed strain; WT, wild-type).

Sanger sequence analysis
We studied a clinical strain of MRSA isolated from a patient. Given that this was a clinical strain, there was a possibility that the MRSA isolates may have undergone mutations. To verify this, the WT isolate was sent for Sanger sequencing, and the resulting sequence was compared with the NCBI database of relevant resistance genes. This process confirmed that the WT isolate had no mutations. Next, the sequence results for the KO isolates were compared with the WT sequence. The Sanger sequencing results were analyzed against the NCBI gene bank (Accession numbers: BA000017, AP003367.1, and D67075.1), and BLAST analysis was conducted. The mutations identified are shown in Figure 5. Sanger sequencing revealed the presence of point mutations and small insertions or deletions (indels) in the target regions of the mecA, aacA, grlA, and grlB genes. These mutations were likely caused by the error-prone nonhomologous end joining (NHEJ) repair mechanism following the Cas9-induced double-strand breaks. Complete gene deletions were not observed; instead, the mutations introduced frameshifts or premature stop codons that disrupted gene function.
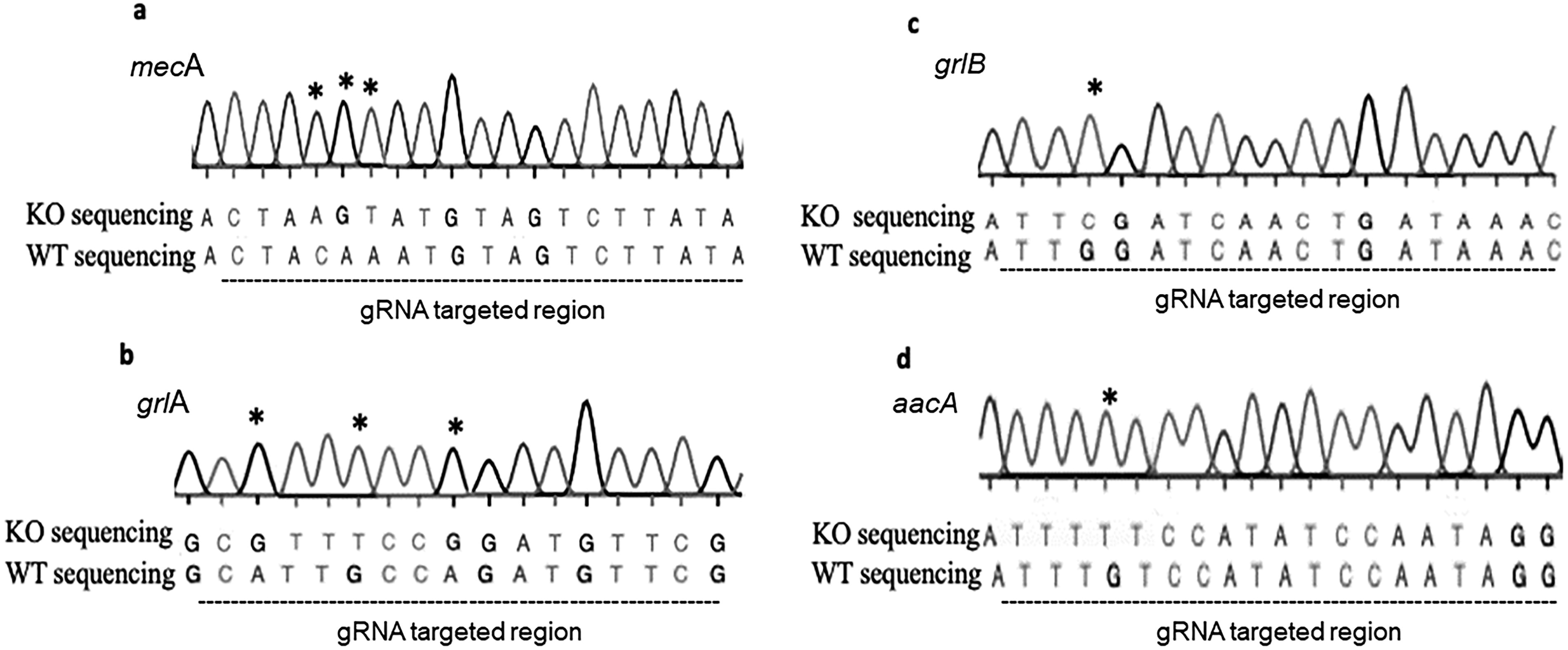
Mutations detected in KO isolate.
Discussion
Antibiotic resistance remains a critical global health and socio-economic challenge, with an increasing urgency to address the growing threats it poses. Beyond the issue of resistance, the selectivity of antibiotics should also be considered. Current antibiotics target essential bacterial functions such as cell wall synthesis, membrane function, protein synthesis, or nucleic acid synthesis. However, these mechanisms are not specific to pathogenic bacteria, often affecting the host’s beneficial microbiota as well. As a result, antibiotics disrupt not only the targeted pathogens but also the host’s normal microbiota.
The stagnation in new antimicrobial development, coupled with the rising rates of antibiotic resistance, highlights the need for innovative therapeutic strategies. CRISPR-Cas technology has emerged as a promising tool for combating antibiotic resistance, offering programmable, sequence-specific antimicrobial interventions. Over the past decade, CRISPR-Cas systems have gained attention as potential antimicrobials capable of targeting specific genes. 31 CRISPR-Cas systems show promise for bacterial genome editing, offering programmable and high-throughput functional genomics with potential applications in tool development, genome homeostasis, and DNA repair. 32 This unique specificity enables CRISPR-based antimicrobials to eliminate antibiotic resistance genes while sparing the beneficial microbiota. The pathogen-specific nature of CRISPR-based genome editing presents considerable potential for advancing CRISPR antimicrobials. CRISPR-Cas systems have the potential to facilitate the selective and quantitative removal of individual bacterial strains, which could in turn enable the development of “smart” antibiotics that evade multidrug resistance and make a distinction between pathogenic and beneficial microorganisms. 33 Various studies have demonstrated that CRISPR machines can be delivered to bacterial populations via vectors such as phages and plasmids to eliminate resistance genes and resensitize bacteria to antibiotics. 34 Early studies on CRISPR antimicrobials aimed to restore bacterial sensitivity to antibiotics by removing resistance genes without harming the normal microbiota. For example, Citorik et al. targeted resistance genes in carbapenem-resistant Enterobacteriaceae and enterohemorrhagic E. coli using phagemids. 35 In contrast, we targeted antibiotic resistance genes in MRSA using the pCasSA plasmid. Both studies aimed to explore the potential of CRISPR-Cas technology in addressing antibiotic resistance, but while Citorik et al. focused on selectively killing resistant bacteria, our study focused on resensitizing MRSA to conventional antibiotics. Yosef et al. similarly suggested targeting beta-lactamase genes in E. coli using phages and Cas3 to prevent resistance gene transfer between strains. 36 In our study, we targeted the mecA, aacA, grlA, and grlB genes in MRSA via Cas9 and plasmid delivery, achieving double-strand breaks in the DNA. These breaks were repaired through error-prone NHEJ, leading to point mutations and small indels in the targeted regions.
Kim et al. developed a system called Re-Sensitization to Antibiotics (ReSAFR) that targeted beta-lactam resistance genes in extended-spectrum beta-lactamase-producing E. coli using CRISPR-Cas9. 37 They used a similar plasmid delivery method to ours but did not perform gene expression or protein analysis. Our study delves deeper, evaluating both genotypic and phenotypic changes through qPCR, western blotting, and susceptibility testing, thus providing a more detailed understanding of antibiotic resensitization. MRSA infections, characterized by their resistance to multiple antibiotics, demand innovative treatment methods. In this study, we designed sgRNAs to target mecA, aacA, grlA, and grlB resistance genes in MRSA. These sgRNAs were cloned into the pCasSA vector, allowing for simultaneous targeting of multiple genes. Successful multiplex gene editing was confirmed through sequencing and further validated by qPCR and western blot analysis, which showed reductions in gene expression and protein levels. Our approach presents a significant advancement over previous studies that primarily focused on single-gene editing. This approach offers a significant advancement over previous studies that focused on single-gene editing or used different CRISPR systems.38,39 The ability to disrupt multiple resistance genes concurrently presents a novel strategy for combating multidrug-resistant pathogens. Compared with the CRISPR-Cpf1 system used by Wang et al. (2022), our Cas9-based approach demonstrates robust efficiency for targeting multiple genes concurrently, with potential applications in bacterial genome engineering. 18 Our study builds on the work of Bikard et al., who targeted mecA and aph-3 in MRSA using phagemid-delivered CRISPR-Cas9. 38 While we share the goal of suppressing antibiotic resistance genes, our method involves plasmid-based delivery, which allowed for the simultaneous targeting of multiple genes. It was observed that mecA and aph-3 gene expression decreased significantly in MRSA strains, while phage immunization occurred in avirulent strains. This immunity will prevent the acquisition of these extrachromosomal structures through the CRISPR system in cases where the strains will acquire antibiotic-resistant genes in the future. 38 In our study, a 30% suppression rate was achieved, leading to a substantial decrease in gene expression and consequent phenotypic shifts in antibiotic susceptibility. Our methodology, which uses plasmid-based delivery and targets multiple genes simultaneously, has differed from the work of Bikard et al. in terms of differences in delivery systems and target specificity. Wang and Nicholaou (2017) used a CRISPR-dCas9 system to suppress mecA gene expression, achieving a 77% decrease. In contrast, our study used active Cas9 to disrupt multiple resistance genes, including aacA, grlA, and grlB. While both approaches achieved significant reductions in gene expression, our findings extend beyond gene expression to include substantial phenotypic changes, such as decreased MIC values and increased susceptibility in disk diffusion assays. 40 Guan et al. (2017) targeted mecA using CRISPR-Cas9, resulting in cytotoxicity, whereas our plasmid-based approach differed in efficiency and stability. 39
Kang et al. (2017) introduced a novel CRISPR nanoparticle system to target mecA, and while we employed plasmid delivery, our study similarly observed phenotypic shifts, emphasizing the reduction in MIC values. 41 Kiga et al. (2020) used Cas13a for sequence-specific bactericidal effects, targeting different resistance genes. 42 In comparison, our study concurrently targeted mecA, grlA, grlB, and aacA, showing both reductions in gene expression and phenotypic shifts. To ensure the specificity of our CRISPR-Cas9 targeting, in silico analysis using Benchling software predicted potential off-target effects. Although no significant off-target sites were predicted, future whole-genome sequencing will be conducted to validate this and ensure the safety of our multiplex gene editing approach.
Our study focuses on mecA, the gene encoding PBP2a, a critical factor in beta-lactam resistance in S. aureus. PBP2a’s low affinity for beta-lactams allows cell wall synthesis to continue despite the presence of these antibiotics, conferring resistance.43,44 Mutations in the mecA gene reduced its mRNA transcription, leading to truncated or non-functional proteins. As a result, the synthesis of functional PBP2a was diminished, restoring the bacteria’s susceptibility to beta-lactams. 45 Without sufficient PBP2a, the bacteria’s cell wall synthesis is hindered in the presence of these antibiotics, rendering the bacteria susceptible once again. 23 The decrease in PBP2a levels was confirmed through western blot analysis, showing a significant reduction in protein expression in the knockout strains compared to the WT strains. This phenotypic change directly correlates with the observed increase in antibiotic susceptibility. In summary, the modification of the mecA gene through CRISPR-Cas9 directly affects the production and functionality of the PBP2a protein. By disrupting the mecA gene, the synthesis of PBP2a is significantly diminished, which in turn restores the bacteria’s susceptibility to beta-lactam antibiotics. This mechanistic insight underscores the efficacy of CRISPR-Cas9 as a precise tool for combating antibiotic resistance.26,38
In conclusion, our study represents a pioneering effort to suppress multiple resistance genes in clinical MRSA isolates using CRISPR-Cas9. The successful elimination of methicillin, ciprofloxacin, and gentamicin resistance, combined with reductions in gene and protein expression, underscores the transformative potential of CRISPR-based therapies in the fight against antibiotic resistance.
Conclusions
The quest for innovative treatments to combat antibiotic resistance has led to extensive research into CRISPR-Cas antimicrobials. Our study, aligned with other literature, highlights the transformative potential of CRISPR-Cas technology in resensitizing resistant bacterial populations. CRISPR technology, with its precise and cost-effective modifications, holds the potential to revolutionize treatment protocols by simultaneously targeting multiple genes. Future research may explore the combination of CRISPR antimicrobials with traditional antibiotics, overcoming limitations related to resistance development. The synergistic effects of phage cocktails containing CRISPR-Cas antimicrobials and nanopharmaceuticals also offer exciting possibilities. CRISPR technology has the potential to reshape treatment paradigms, positioning itself as a key tool in the fight against antibiotic resistance.
Footnotes
Acknowledgments
Authors’ Contributions
A.A., C.T., and Ş.E. designed the experiments. A.A. performed microbiology and molecular biology experiments. A.A., C.T., and Ş.E. analyzed the data and wrote the article. All authors read and approved the final version of the article.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This research was supported by the Ege University Scientific Research Projects Coordination by Grant number TDK-2020-21898.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.