
Abstract
Treating human genetic conditions in vivo requires efficient delivery of the CRISPR gene editing machinery to the affected cells and organs. The gene editing field has seen clinical advances with ex vivo therapies and with in vivo delivery to the liver using lipid nanoparticle technology. Adeno-associated virus (AAV) serotypes have been discovered and engineered to deliver genetic material to nearly every organ in the body. However, the large size of most CRISPR-Cas systems limits packaging into the viral genome and reduces drug development flexibility and manufacturing efficiency. Here, we demonstrate efficient CRISPR gene editing using a miniature CRISPR-Cas12f system with expanded genome targeting packaged into AAV particles. We identified efficient guides for four therapeutic gene targets and encoded the guides and the Cas12f nuclease into a single AAV. We then demonstrate editing in multiple cell lines, patient fibroblasts, and primary hepatocytes. We then screened the cells for off-target editing, demonstrating the safety of the therapeutics. These results represent an important step in applying CRISPR editing to diverse genetic sequences and organs in the body.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) systems are versatile tools for targeted gene editing.1–6 The clinical promise of CRISPR-Cas technology recently took a major step forward with the approval of Casgevy as an ex vivo gene editing treatment for sickle cell disease. 7 Several clinical trials are underway using gene editing as a tool to create ex vivo therapies for hematologic diseases and cancers (CAR-T). 8 Delivering the Cas editing machinery directly to patients through in vivo therapies is also advancing with clinical trials using lipid nanoparticles (LNPs) to deliver editors to the liver. 9 A major hurdle for LNP therapies is the lack of tissue distribution, with most LNP formulations restricted to delivery to the liver. 10
By contrast, natural and synthetic adeno-associated virus (AAV) serotypes have been developed that target many tissues in the body, including the liver, lungs, eye, heart, muscles, kidneys, and the central nervous system (CNS). 11 Several gene therapies using AAV have been approved by the Food and Drug Administration (FDA) to treat diseases of the liver, eye, and CNS. 12 Safe and effective AAV delivery of therapeutic gene editing cargo would therefore broaden the spectrum of accessible tissues.
The biggest limitation to delivering editing machinery by AAV is the relatively small genetic payload (4.5 kb). 13 The most widely used CRISPR-Cas systems (Cas12a and Cas9) are large proteins ranging from 120 to 160 kDa (3.2–4.3 kb) and require additional fusions like nuclear localization signals for effective editing in human cells.5,14 As AAV delivers a genetic payload, the cargo must also include the requisite promoters and terminators for transcription and translation as well as separate promoters for the single guide RNA (sgRNA). Splitting the genetic cargo into multiple vectors or using minimal promoters with the smaller Cas nucleases is an imperfect solution to this problem and requires significant tradeoffs in AAV packaging, genetic stability, payload expression, and drug development.15,16
The recent discovery of the highly compact CRISPR-Cas12f nuclease family (60 kDa) poses an elegant solution to delivery of editing machinery using AAV.17–25 The Cas12f family functions as a protein dimer around a sgRNA, forming an editing complex that is equivalent to the larger Cas9 and Cas12a families but encoded in half the genetic space. 26 We previously identified a novel set of Cas12f proteins and engineered them for editing in human cells, including AI1Cas12f1, a 486-amino-acid protein from the genus Alistipes with a preferred NTTR PAM sequence. 21 In this work, we develop AAV gene editing therapies encoding Al1Cas12f1 along with guides for several in vivo therapeutic targets and demonstrate editing and off-target analysis in cell lines and primary cells. This work is a critical step toward an effective and efficient AAV gene editing therapy.
Materials and Methods
Plasmid construction and AAV vector production
Nuclease and sgRNA plasmids used in transfection experiments were constructed as described previously. 21 For AAV production, a codon-optimized Al1Cas12f1 gene was synthesized with an N-terminal SV40 nuclear localization sequence (NLS) and C-terminal nucleoplasmin NLS followed by a 3× human influenza hemagglutinin (HA) tag (Twist Biosciences). The Al1Cas12f1 sequence was inserted downstream of a CMV promoter in sgRNA expression plasmids containing inverted terminal repeat sequences. AAV vectors were produced by BPS Bioscience, yielding 1.1–3.6 × 1012 viral genomes per milliliter.
Cell culture and plasmid transfection
HEK293T (CRL-3216, ATCC) and NIH3T3 cells (CRL-1658, ATCC) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Fibroblasts from patients with spinal muscular atrophy (SMA) were maintained in EMEM supplemented with 10% FBS, 1% penicillin–streptomycin, and 1× GlutaMAX (ThermoFisher). Mouse primary hepatocytes (Cell Biologics C57-6224F) were thawed in rodent hepatocyte thawing medium (Lonza MCRT50), plated with hepatocyte plating medium (Lonza MP100), and maintained in hepatocyte maintenance medium (Lonza MM250) according to the manufacturer’s recommendations. All cells were kept at 37°C in an incubator with 5% CO2. Nuclease and sgRNA plasmids were cotransfected into HEK293T cells using Mirus Transit X2 reagent. Experiments were conducted in 96-well plates seeded with 5 × 104 cells/well and transfected with 100 ng nuclease expression vector and 100 ng sgRNA expression vector according to the manufacturer’s recommendations. Samples were incubated for 72 h.
Al1Cas12f1-AAV-DJ transduction
Cells were seeded at 2–3 × 104 cells/well into 96-well plates 24 h prior to AAV transduction at 100k MOI. HEK293T, mouse primary hepatocyte, and NIH3T3 experiments were incubated for 4–7 days with media changes every 3 days. Mouse primary hepatocyte medium was exchanged every day according to Lonza recommendations. Fibroblasts from patients with SMA were treated with 60 µM etoposide 5 h after seeding. Etoposide-containing medium was removed at 24 h, immediately prior to AAV transduction. The SMA patient fibroblast experiment was incubated for 13 days and medium was changed every 3 days.
Genomic DNA extraction and sequencing
Genomic DNA was harvested with QuickExtract (LGC Biosearch Technologies) and amplified using KAPA HiFi polymerase with genomic site-specific primers (Supplementary Table 3). Amplicons from plasmid-transfected samples were submitted for Sanger sequencing, and editing efficiency was determined with TIDE analysis. 27 Amplicons from AAV-transduced samples were sequenced via Illumina iSeq with 150 bp paired-end reads, and indel frequency was quantified with CRISPResso2. 28 All editing data were plotted using Prism software.
Off-target prediction
A custom version of Cas-OFFinder 29 was used to search for off-targets related to PRSS1-g11 and SMN2-g1 guides in the human genome and PCSK9-g1 in the mouse genome (Supplementary Table 2). A DTTR (D = A, G, or T; R = A or G) PAM was used for Al1Cas12f1. 21 For PRSS1-g11 and PCSK9-g1, all predicted off-targets where the combined DNA/RNA bulge size and number of mismatches totaled 4 or less were selected. For SMN2-g1, all predicted off-targets without bulges and <4 mismatches or <3 bulge size with one mismatch were selected. There were many predicted off-targets with two mismatches and a bulge size of 1, so two were selected that had preferred PAMs. Off-target sites were amplified and sequenced via Illumina iSeq as described earlier. Raw FASTQ files can be accessed at BioProject PRJNA1093476 in the Sequence Read Archive (SRA), NCBI.
Results
Al1Cas12f1 edits therapeutic targets via plasmid transfection
The PRSS1 gene encodes cationic trypsinogen and is dysregulated in patients suffering from hereditary pancreatitis. 30 We designed 12 guides for Al1Cas12f1 targeting the exons of PRSS1 to disrupt trypsinogen production (Fig. 1A, Supplementary Table 1). sgRNA expression vectors were cotransfected with an Al1Cas12f1 expression vector and assessed for PRSS1 editing in HEK293T cells. All guides exhibited PRSS1 editing, with PRSS1-g11 having the highest efficiency (Fig. 1B). PRSS1-g11 targets exon 5 and has an ATTA PAM, which is preferred by Al1Cas12f1. 21

Guide selection for PRSS1 and SMN2.
Disruption of the ISS-N1 region downstream of SMN2 exon 7 is a proven therapeutic technique for treatment of SMA. 31 We designed two guides with cut sites in this 15-nt-long ISS-N1 region (Fig. 1C). Guides were tested for editing with Al1Cas12f1 and Un1Cas12f1 19 (Fig. 1D). Un1Cas12f1 was unable to efficiently edit SMN2 with either guide. By contrast, Al1Cas12f1 was capable of effective editing with SMN2-g1. The higher editing with Al1Cas12f1 on SMN2-g1 is likely owing to the preference for ATTA PAM sequences, whereas this PAM sequence is not preferred by Un1Cas12f1 (TTTN PAM).21,32
Al1Cas12f1 efficiently edits therapeutic targets in human and mouse cells via AAV delivery
Al1Cas12f1 was packaged into AAV-DJ vectors 33 with the top sgRNAs targeting human genes PRSS1 and SMN2, as well as mouse genes PCSK9 and TTR (Fig. 2A, Supplementary Figure 1). PCSK9 and TTR are therapeutic liver targets for treating familial hypercholesterolemia and ATTR amyloidosis, respectively.9,34 PRSS1 and SMN2 targeting vectors were transduced into HEK293T cells, yielding ∼40% editing efficiency (Fig. 2B). In addition, the SMN2 targeting construct was transduced into fibroblast cells from patients with SMA, yielding ∼10% editing efficiency. Efficient editing of the mouse targets (>5%) could not be observed through plasmid transfection, likely owing to poor plasmid transfection of NIH3T3 cells (Supplementary Fig. S2). As we were unable to select top performing guides, we decided to package multiple guides for PCSK9 and one for TTR directly into AAV for testing. PCSK9 and TTR targeting vectors were transduced into NIH3T3 mouse cells and mouse primary hepatocytes. A maximum of ∼40% editing efficiency was reached for PCSK9 in mouse primary hepatocytes, whereas TTR editing reached ∼10% (Fig. 2C).
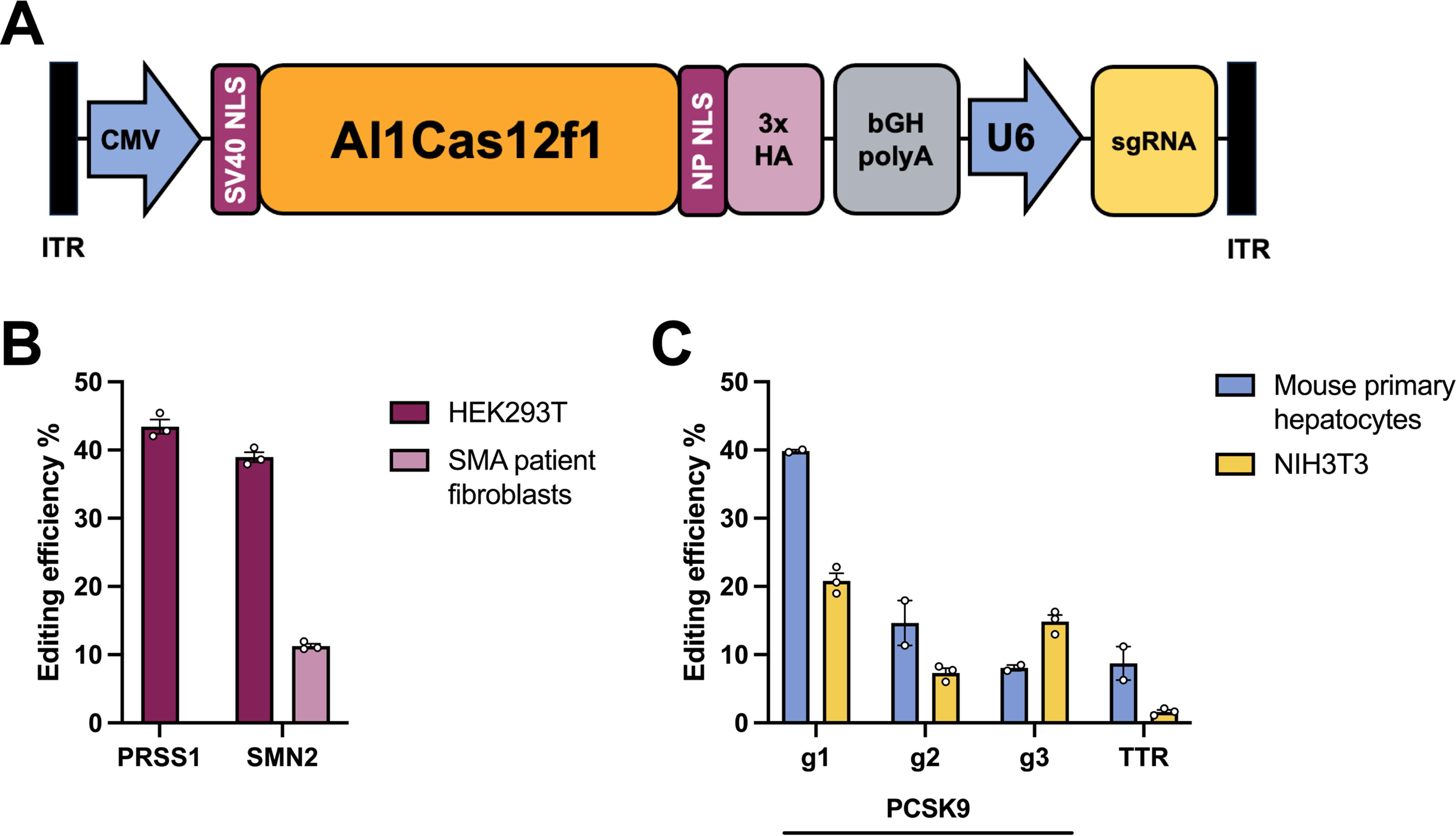
Al1Cas12f1 editing via AAV transduction.
Low off-target editing with Al1Cas12f1-AAV-DJ
To assess the specificity of our novel Cas12f AAV construct, predicted off-targets were tested for top PRSS1, SMN2, and PCSK9 guides. Off-target sites with the fewest mismatches and DNA/RNA bulges were amplified and sequenced from samples of the highest edited cell type. For PRSS1-targeting AAV, the highest off-target editing was 1.3% for a two-mismatch intergenic off-target site located between the PRSS1 and PRSS2 genes (Fig. 3A). For SMN2-targeting AAV, the highest off-target editing was 0.3% for an intronic off-target with one mismatch and a 2-bp RNA bulge located in the DGKI gene (Fig. 3B). For PCSK9 in mouse primary hepatocytes, the highest off-target was under 0.1% editing efficiency, with two mismatches and a 1-bp RNA bulge (Fig. 3C).

Editing of predicted off-target sites. Off-target sites were predicted in silico for
Discussion
The approval of the first CRISPR gene editing therapy highlights the tremendous potential for the technology to benefit human health. 7 Advancing CRISPR technology to treat patients in vivo requires optimizing the delivery of the CRISPR-Cas editing machinery to patient tissues. AAV particles are a proven in vivo delivery technology used in multiple FDA-approved gene therapies. 11 A major hurdle for AAV therapies is the physical limit for viral genome size. DNA payloads that are near this limit have decreased packaging efficiency and result in empty AAV particles. 35 This precludes efficient packaging of many CRISPR-Cas systems such as Cas9 or Cas12a. The CRISPR-Cas12f family is unique owing to their very small size, roughly half of most CRISPR-Cas nucleases. Despite their small size, the enzyme are fully active editors that function using a dimeric structure that is based around a sgRNA.21–25 The compact size of Cas12f nucleases allows for the inclusion of full-length promoters, guides, and other genetic elements that increase the efficiency and efficacy of AAV therapies.
In this work, we take advantage of the small size and broad PAM recognition of the recently discovered Al1Cas12f1 enzyme 21 to package complete editing vectors in AAV particles. We first selected therapeutic gene targets (PCSK9, TTR, SMN2, and PRSS1) that are addressable in vivo using AAV delivery. We then screened multiple guides for each gene using plasmid transfection of HEK293T or NIH3T3 cells to identify the guides with the highest editing potential. Multiple guides for PCKS9 and TTR were used for further studies owing to the low editing observed with plasmid transfection. For the human gene targets, the best performing guides contained an ATTA PAM, the preferred PAM for Al1Cas12f1. 21
We then designed AAV payloads encoding the complete Al1Cas12f1 sequence driven by the full-length CMV promoter and sgRNA driven by the U6 promoter. The payloads were packaged into the AAV-DJ serotype and evaluated for therapeutic gene editing in human or mouse cells. The SMN2- and PRSS1-targeting AAV constructs efficiently edited human HEK293T cells and primary SMA patient fibroblasts (SNM2 only). AAV targeting PCSK9 and TTR effectively edited mouse primary hepatocytes as well as the mouse NIH3T3 cell line. These results demonstrate both the editing efficiency of Al1Cas12f1 and the ability of the nuclease and guide to be efficiently packaged in AAV particles. Off-target analysis of edited cells shows relatively low levels of editing an untargeted site; however, more thorough analysis will be needed to advance these therapies. Together, these data demonstrate the efficiency and utility of Al1Cas12f1 for gene editing applications.
Footnotes
Acknowledgments
The authors thank Prof. Joseph Bondy-Denomy for critical reading of the article.
Authors’ Contributions
A.S. and L.A.T. contributed to the study design, experiments, data analysis, and article writing. Z.M. and J.S.-A. contributed to experiments and data analysis. T.C. and D.R. contributed to study design. M.S. contributed to study design, data analysis, and article writing.
Author Disclosure Statement
All authors are employees of Acrigen Biosciences, Inc. A patent was filed relating to some of the findings presented in this study.
Funding Information
This work was supported by the National Science Foundation (2136335) to DR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.