
Abstract
Flax is an important crop used for oil and fiber production. Although genetic engineering has been possible in flax, it is not commonly used to produce cultivars. However, the use of genome editing technology, which can produce site-specific mutations without introducing foreign genes, may be a valuable tool for creating elite cultivars that can be easily cultivated. The purpose of this study was to investigate the potential of genome editing in flax by establishing the clustered regularly interspaced short palindromic repeats (CR ISPR)-CRISPR-associated protein 9 (CRISPR-Cas9) genome editing system using the phytoene desaturase (PDS) gene, which produces albino mutants that are easily identifiable. Four sgRNAs were designed from two PDS genes of Flax (LuPDS1 and LuPDS2), and CRISPR-Cas9 genome editing vectors were constructed. After gene transformation, albino phenotypes were observed in transformed callus and regenerated plantlets on selection media. Polymerase chain reaction (PCR) amplification and sequencing of the PDS genes revealed deletions and insertions in the albino tissues, indicating successful editing of the PDS genes. Potential off-target sites were analyzed, but no off-target mutations were found, indicating the specificity of the CRISPR-Cas9 system. The establishment of a flax genome editing system using the CRISPR-Cas9 technology opens up new possibilities for the genetic engineering of flax. This study demonstrates the potential of genome editing in creating elite cultivars that can be easily cultivated, which can have significant implications for the flax industry.
Introduction
Flax (Linum usitatissimum L.) is an annual crop belonging to the genus Linum in the Linaceae family. Flax is economically important for providing fibers for clothes and edible oil for human consumption. Flaxseed oil has beneficial functions in regulating coronary heart disease, reducing hormonal and neurological disorders, and reducing the risk of cancers.1–5
Genetic engineering gives opportunities to improve the yield and quality of flax more precisely and efficiently than traditional breeding methods. Genetic modification has been applied in flax, including improvement of resistance to herbicides and diseases,6,7 quality of flax seeds,8,9 and resistance to heavy metals. 10 In recent years, the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (CRISPR-Cas9) technology has been widely used in many important economic crops due to its simple design, accurate and efficient generation of mutations, and easy separation of introduced foreign DNAs to make transgene-free plants. 11 However, an efficient genome editing system in flax remains to be established.
PDS gene has been reported as a marker indicator for CRISPR-Cas9-mediated gene editing in several plant species, including apple, 12 grapes, 13 cassava, 14 Populus, 15 Arabidopsis thaliana, 16 Citrus sinensis, 17 Medicago truncatula, 18 and Petunia hybrid. 19 Mutations causing defects in phytoene desaturase (PDS) result in plant albinism and dwarfing. In this study, we identified two flax PDS genes encoding the rate-limiting enzymes involved in carotenoid biosynthesis. They were chosen as the target genes to establish a CRISPR-Cas9 genome editing system in flax.
Two binary vectors containing four sgRNAs targeting the PDS genes were designed and co-transformed to flax through Agrobacterium-mediated gene transformation, albino transgenic calli, and plantlets were observed indicating possible mutation of the PDS genes. The PDS genes of the albino calli and plantlets were further analyzed by DNA sequencing to confirm the editing of the PDS genes. Our results indicate that the CRISPR-Cas9 system is sufficient to create mutations in flax, and has the potential for flax improvement.
Materials and Methods
Plant materials
Flax plants for our study from the Zhangjiakou Agricultural Science Academy. The plants used in this study were grown in a growth chamber at 22°C under a 16/8-h photoperiod light cycle. Young leaves were collected from individual plants after 6 weeks of growth and immediately frozen in liquid nitrogen. The plant samples were then stored at −70°C.
Cloning of LuPDS genes
Genomic DNA was isolated from flax young leaves by using the Mini BEST Plant DNA Extraction Kit (TaKaRa) according to the manufacturer’s recommendation. Genomic DNA fragments of the two flax PDS genes LuPDS1 and LuPDS2 (glus10021967 and glus10041260) were amplified with two pairs of primers gLuPDS1F/gLuPDS1R ATGTACGGGAGCGCCACCGTCT; CTACTAGCTTTGAACAGCTTAC) and gLuPDS2F/gLuPDS2R (ATGTACGGGAGCGCCACCGT; CTACTAGCTTTGAACAGCTTACT), respectively designed based on the flax genome sequence (https://phytozome-next.jgi.doe.gov/). The PCR amplification was completed with PrimeSTAR® Max DNA Polymerase (Takara) in a total volume of 50 µL, with the following reaction conditions: 5 min template denature at 95°C, 35 cycles of 95°C denature for 30 s, 55°C annealing for 30 s, 72°C extension for 1 min, followed by 72°C extension for 5 min after the cycling. The PCR products were purified with a DNA cleaning kit (Beyotime, China), and cloned into the TA/Blunt-Zero vector (Vazyme, China) according to the manufacturer’s recommendation. Cloned PCR products were analyzed by Sanger sequencing (Sangon Biotech, China).
Construction of CRISPR vectors for genome editing
The two LuPDS gene sequences were aligned by DNAMAN software (Lynnon Biosoft). Four target sites in two conserved regions were designed (Fig. 1a). Target 1 (LuPDS22) and Target 2 (LuPDS57) were located in the first exon of the LuPDS genes, while target 3 (LuPDS366) and target 4 (LuPDS398) were located in exon 3 (Fig. 1b). Specific primers containing the gRNAs for the four target sites were designed and synthesized (Table 1).
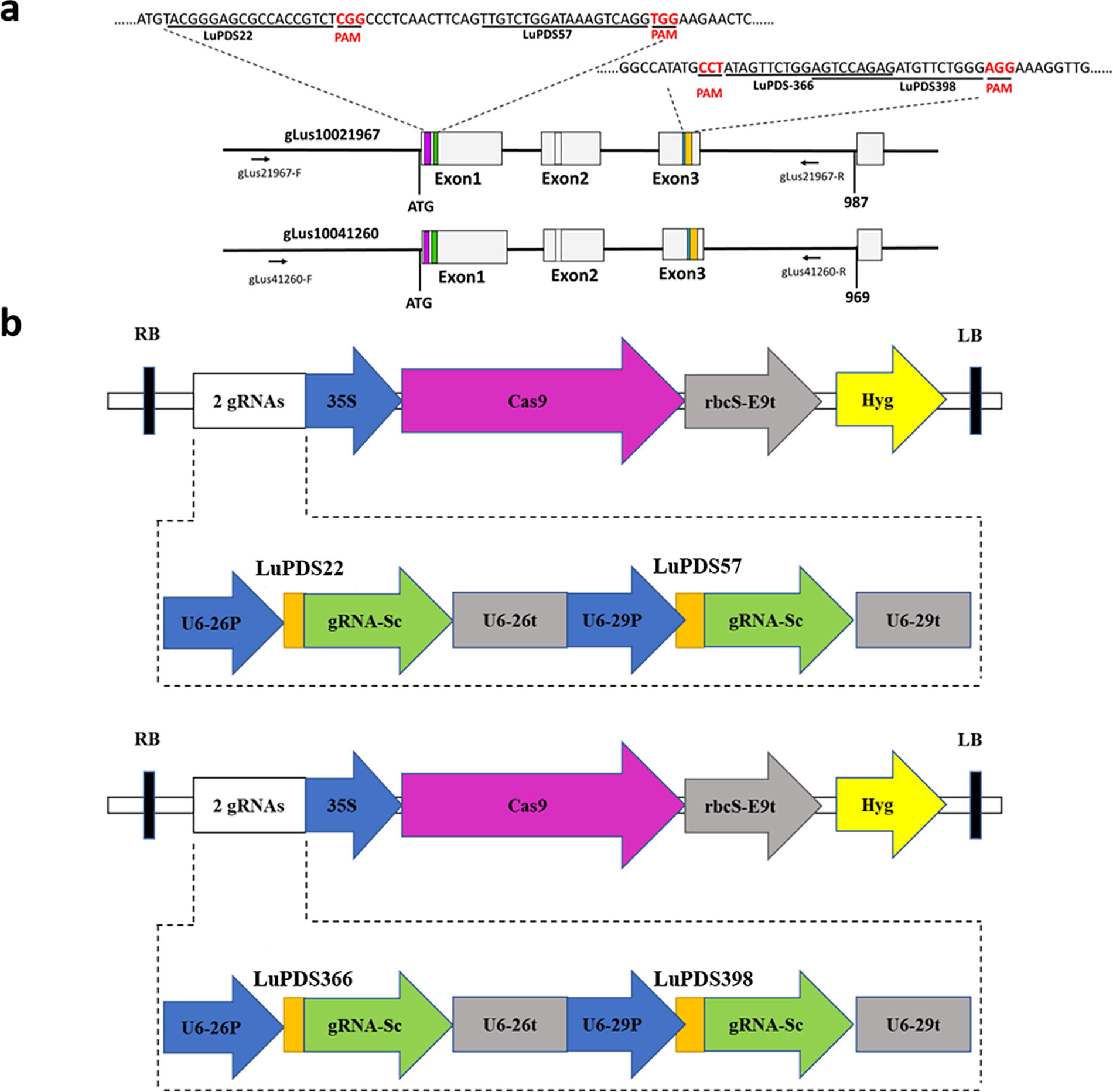
Plasmid constructs for genome editing of LuPDS.
gRNA Primers
To assemble two gRNAs into the PHSE401 vector, a pair of primers containing the target sites in the forward and reverse primers (LuPDS_S22-BsF and LuPDS_S57-BsR for LuPDS22 and LuPDS57; LuPDS_S-366-BsF and LuPDS_S398-BsR for LuPDS366 and LuPDS398), respectively were used to amplify PCR fragments in a PCR reaction with the pCBC-T1T2 plasmid DNA as the template. The PCR amplification was completed with PrimeSTAR® Max DNA Polymerase (Takara) in a total volume of 50 µL, with the following reaction conditions: 95°C template denature for 5 min, 35 cycles of 95°C denature for 30 s, 55°C annealing for 30 s, 72°C extension for 1 min, followed by 72°C for 5 min after the cycling. The double-stranded DNA fragments (T1T2-PCR) were firstly purified, digested with BsaI, and ligated into the pHSE401 vector by using the T4 Ligase (New England Biolabs) to generate two final LuPDS CRISPR-Cas9 vectors (pCAMBIA-35S::Cas9-AtU626p::sgRNA1-AtU629p::sgRNA2 and pCAMBIA-35S::Cas9-AtU626p::sgRNA3-AtU629p::sgRNA4) (Fig. 1c). The digestion and ligation reaction contains 2 µL PCR fragment, 2 µL pHSE401 vector, 1.5 µL 10xNEB T4 Buffer, 1.5 µL 10xBSA, 1 µL BsaI (NEB), 1 µL T4 Ligase (NEB), and 6 µL ddH2O in a total volume of 15 µL. The reaction was performed at 37°C for 5 h, followed by 5 min at 50°C and 10 min at 80°C. All these constructed vectors were transformed into Escherichia coli and verified by sequencing (Sangon Biotech, China). Then, the validated pHSE401-2gR-LuPDS recombinant plasmids (pCAMBIA-35::Cas9-AtU626p::sgRNA1-AtU629p::sgRNA2 and pCAMBIA-35S::Cas9-AtU626p::sgRNA3-AtU629p::sgRNA4) were transformed into Agrobacterium (strain EHA105) cells for further transfection of flax explants respectively.
Flax transformation
Cultivated flax seeds were surface sterilized by soaking in 75% alcohol for 2 min, followed by twice 20 min sterilization in 10% sodium hypochlorite solution. After 7–8 times rinsing with sterile water, flax seeds were dried on sterile filter paper and sown on 1/2 MS medium (2.247 g/L MS +10 g/L Sucrose +9 g/L Agar, pH 5.7). The seeds were germinated at 22°C in a growth chamber under dark conditions for 3 days and then transferred to light conditions to grow for 4 days at 22°C. The hypocotyls of 7-day-old flax seedlings were harvested as explants for genetic transformation.
Hypocotyls were cut aseptically into 0.5–1 cm sections as explants for transformation. The explants were inoculated by swirling in the two mixes of Agrobacterium tumefaciens EHA105s containing pHSE401-2gR-LuPDS recombinant plasmids for approximately 2 h. Then the infected hypocotyl explants were co-cultivated in differentiation medium (4.43 g/L MS + 0.1 mg/L NAA + 1 mg/L 6-BA + 100 mg/L AS + 30 g/L Sucrose + 9 g/L Agar, pH 5.7) for 3 days at 20°C in the dark. After co-cultivation, the infected hypocotyl explants were washed 3 times with sterilized distilled water containing 200 mg/L cefotaxime and then transferred into the selective medium (4.43 g/L MS + 0.1 mg/L NAA + 1 mg/L 6-BA + 400 mg/L Cef + 25 mg/L HyB + 30 g/L Sucrose + 9 g/L Agar, pH 5.7). The infected hypocotyl explants were sub-cultured every 2 weeks to select transgenic plants.
Detection of mutation
Genomic DNAs were extracted from wild type flax and transgenic plants selected on the selection medium by using a Mini BEST Plant DNA Extraction Kit (TaKaRa). To analyze the mutations in the target region, primers (pLuPDS-F: TTGTCATGGAACGAACGATGCTC; pLuPDS-R: TGACATTAAACTTAGTACAATCTAAG) were designed to amplify a DNA fragment flanking the four targets. The PCR amplification was completed with Prime STAR® Max DNA Polymerase (Takara) in a total volume of 50 µL, with the following reaction conditions: 95°C denature for 5 min, 35 cycles of denature at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min, followed by annealing at 72°C for 5 min after the cycling. The PCR products were purified and cloned into the TA/Blunt-Zero vector (Vazyme, China), then single colonies were selected and analyzed by sequencing. Sequences from the cloned PCR products were aligned with wild type sequences using DNAMAN to detect mutations. Ten single clones from each stable transgenic line with mutants were selected for further analysis to identify the mutation.
Potential off-target analysis
In order to detect CRISPR off-target mutations in the four targets of LuPDS gene. The putative off-target sites were predicted by using the online CRISPOR (CRISPOR.tefor.net) tools. Genomic DNAs were extracted from transgenic plants and wild type. Primers (Supplementary Fig. S1) were designed and used to amplify sequences flanking the off-target sites by PCR. PCR products were cloned and analyzed according to the method described in the previous section.
Statistical analysis
Prism GraphPad software Ver. 5.01 was used for statistical analyses. Firstly, we calculated the mutation frequency at different loci and compared the mutation frequencies between different loci. Then, we performed a paired t-test using Student’s t-test for statistical analysis. The statistical significance was checked at p ≤ 0.01 (*p ≤ 0.01), indicating that the results were considered significant.
Results
Flax PDS gene cloning and analyses
PDS is the rate-limiting enzyme in the carotenoid biosynthetic pathway. PDS mutants often have clearly visible albino and dwarf phenotypes at the juvenile stage. 12 Thus, we selected the flax PDS genes as the target for genome editing by the CRISPR-Cas9 system.
The first three exons of two LuPDS genes were amplified respectively, and then purified from agarose gels, cloned, and sequenced. The sequenced result showed no difference with the sequence in the genome database. Sequence alignment showed high similarity between LuPDS1 and LuPDS2, while only a few small SNPs. On the basis of the conserved sequence between LuPDS1 and LuPDS2, four sgRNAs (LuPDS22, LuPDS57, LuPDS366 and LuPDS398) were designed to target four conserved sites located (Fig. 1a).
Target selection and vector construction
The target site sequence is located in the conserved sequence in the first three exons of the two LuPDS genes. Representatively, two sgRNAs (LuPDS22 and LuPDS57) were designed to target two conserved sequences in the first exon, and another two sgRNAs (LuPDS366 and LuPDS398) were designed to target two conserved sequences in the third exon (Fig. 1a). The CRISPR-Cas9 machinery contained two sgRNAs expression cassettes, activated by U6-29P and U6-26P, respectively (Fig. 1b). DNA fragments containing two target sites were synthesized by a PCR reaction with the pCBC-T1T2 plasmid DNA as the template was cloned into the PHSE401 CRISPR-Cas9 vector for flax transformation. 20
Flax transformation
The genetic transformation methods of flax have been reported in previous studies. 21 In our present study, we established a stable agrobacterium-mediated genetic transformation system for golden flax. We selected the hypocotyls of 7-day-old golden flax as explants. Then the flax explants were infected by the agrobacterium strain EHA105, which carries the target vector. Three stable transformation experiments were performed and resulted in different transformation rates. A total of 500 flax hypocotyls were infected with agrobacterium tumefaciens, among them, 56 callus were acquired from the selective medium containing 5 mg·L—1 hygromycin (Hyg) after 6 months.
CRISPR-Cas9-mediated mutagenesis of LuPDS in transgenic flax
To test whether the CRISPR-Cas9 system can be successfully applied to flax for gene knockout and produce the ideal phenotype, a binary vector of CRISPR-Cas9 machinery carrying two sgRNAs, one cas9 and the Hyg resistance gene, which was transformed into flax hypocotyls explants by Agrobacterium-mediated transformation. The transformed callus regenerated and germinated on the Hyg selection medium. After 2 months, the Hyg-resistant callus with the following two phenotypes, including variegated with both albino and green, normal green (Fig. 2a).
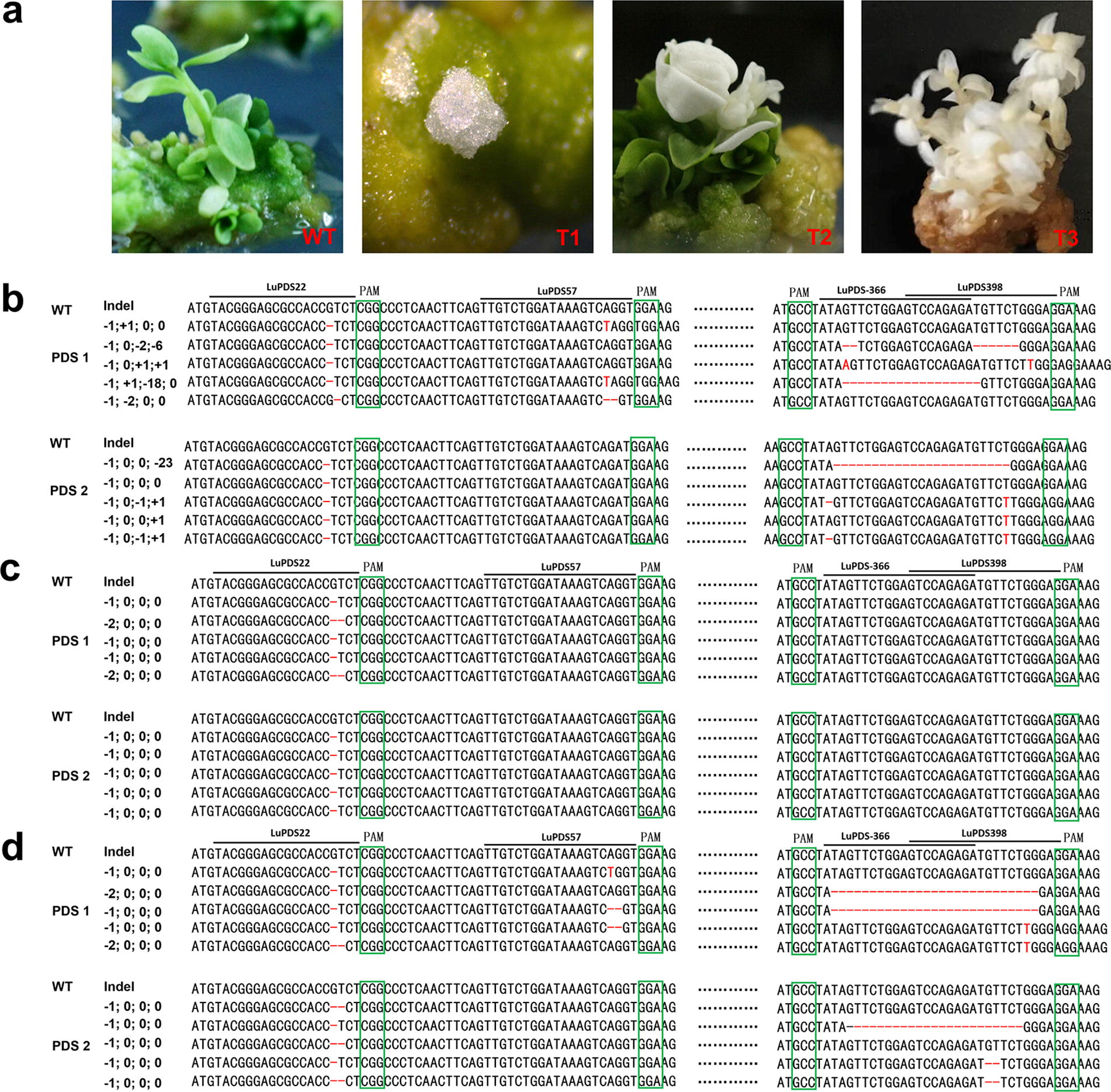
Targeted gene editing in transgenic flax plants using CRISPR-Cas9 system.
In order to test whether the target sequence would be destroyed by CRISPR-Cas9. The PCR was performed on transgenic flax callus and plant by using primers flanking the target region of the two PDS genes. No obvious size change of the amplicons after being examined by gel electrophoresis.
In order to further test the mutation efficiency of various targets and mutation types in the T0 generation, we randomly selected one mosaic white callus (T1), one transgenic mosaic albino plant (T2), and one transgenic albino plant (T3) for PCR and sanger sequencing. In total, 30 single clones from three transgenic lines were sequenced. For the LuPDS22, the PDS1 of callus (T1) and plant (T2, T3) were heterozygous respectively. (Fig. 2), the PDS2 of callus and plant (T2) were monoallelic homozygous with a 1 bp deletion. For the LuPDS57, the PDS1 of callus was heterozygous, carrying an allele with 2 bp deletion and the other allele with 1 bp substitution, while, the mutation of PDS2 in callus and plant, the mutation of PDS1 in plant were not found. In addition, for the LuPDS366, the PDS1 and PDS2 of callus were chimeric, with deletion (−1bp, −2 bp, −18 bp, and −23 bp) and 1 bp insertion. For the LuPDS398, the presence of PDS in the callus was only edited with 1 bp insertion. Furthermore, we found that target 1 (100%) had the highest mutation efficiency, significantly higher (p < 0.01) than the other targets, and the most common mutation in the LuPDS22 site was 1-bp deletion (Table 2).
Mutations in Different Target Sites
deletion span two target sites are counted for both sites.
p < 0.01.
Potential off-target mutation analysis
Four potential off-target mutation sites were detected through CRISPOR (Table 3). Sequencing analysis of 9 independent samples by PCR amplification. The results indicated that no off-target phenomenon occurred in flax (Supplementary Fig. S1).
Putative off-target Sequences
Discussion
Genome editing in Flax with single-stranded Oligo Deoxy Nucleotides was successful by Oligonucleotide-directed mutagenesis (ODM). 22 ODM relies on the introduction of chemically synthesized oligonucleotides and reagents that cause double strand breaks (DSBs). DSBs are created with nontargeted reagents such as the bleomycin family of glycopeptide antibiotics, or targeted reagents such as meganucleases, zinc finger nucleases, transcription activator-like effector nucleases, and CRISPR-Cas9 nuclease system. The introduced oligos may act as DNA templates in DSB repair in a template-directed manner to generate precise genome editing. The ODM technology has been used in bacteria, fungi, animals, and plants.23–28 With the help of the CRISPR-Cas9 site-specific system, Sauer et al. created single-base editing of the EPSPS gene in flax by the ODM technology. 22 Mutations at two sites (T178 and P182) were obtained although the mutation rate was low. They successfully regenerated fertile plants from protoplasts culture and obtained mutants with herbicide resistance. 22 However, the low editing frequency and the difficulty of regenerating mutant plants from protoplasts have prevented the application of this technology in flax improvement. Furthermore, while Saha et al. developed gRNAs for 34 HSF genes involved in the heat stress response, the application of the CRISPR-Cas9 system for gene editing in flax has not been explored. 29 Therefore, our study contributes significantly to the field by presenting a comprehensive protocol for establishing a CRISPR-Cas9-mediated genome editing system specifically tailored for flax.
CRISPR-Cas9 is an efficient tool for genome editing in many plant species, including apple, 30 grape,13,31 and orange. 32 To explore the benefits of CRISPR-Cas9 system in flax genetic engineering, we tried to develop an efficient genome editing system by using the PDS gene. The PDS gene plays an important role in the carotenoid synthesis pathway and is mainly expressed in plant leaves, flowers, and fruits. When its synthesis is blocked, plants will exhibit photobleaching, and the phenotype is easy to observe. 33 Thus, the PDS gene has frequently been used as a marker indicator for CRISPR-Cas9-mediated gene editing. In this report, we successfully edited two LuPDS genes and obtained parameters for flax genome editing. The mutant callus and plantlets showed clear albino phenotypes.
Previous studies reported that multiple targets for one gene can improve mutation efficiency. 20 Here, we designed four targets for PDS genes carried by two binary vectors, after co-transformation, we successfully demonstrated that the CRISPR-Cas9 system can efficiently edit the PDS gene of flax and endow an evident albino phenotype (Fig. 2a), which strongly indicated that CRISPR-Cas9 system was capable of performing genome editing in flax genome.
The efficiency of CRISPR-Cas9 mutation is influenced by many factors, including transformation methods, selective and reporter genes, Cas9 promoter, and sgRNA sequence. 34 Huge differences exist in the regeneration and transformation efficiency of different varieties of flax. The most commonly used method of flax transformation is Agrobacterium infection of hypocotyls. 35 In this study, we established an effective genetic transformation system of flax callus, which laid an important foundation for further screening and obtaining transgenic plants. A previous study reported kanamycin as a selective agent in flax transformation may lead cells to escape. 21 Thus, we selected Hyg as a selective marker in flax transformation, which has been used in previous flax transformations. 36 The U6 promoter of Arabidopis thaliana is the most commonly used promoter to drive sgRNA expression in plant species. 37 In this current study, AtU626 and AtU629 promoters were used to drive the expression of sgRNAs in flax respectively. In addition, the Cauliflower mosaic virus 35S (CaMV35S) promoter has been successfully used in the transformation of flax. 38 Thus, in this study, we used the CaMV35S promoter to drive a maize codon-optimized Cas9 expression in flax. Here, we have successfully obtained the transgenic calli and shoots (Fig. 2a).
As reported in previous studies, CRISPR-Cas9 generated mutations are mainly short insertion and deletion in different plant species, including potato,39,40 poplar, 41 watermelon, 33 and soya bean. 42 These results support our finding that most mutations are 1 bp deletions (Fig. 2b and c). Moreover, we also identified large deletions (18 bp and 23 bp) between target LuPDS366 and LuPDS398. These results indicate that CRISPR-Cas9 system can be used for accurate genome editing of the first generation of flax.
To test whether the GC content in the target sequence results in a high editing efficiency, we designed four targets with different GC content including LuPDS22 68.42%, LuPDS57 42.11%, LuPDS366 47.37%, LuPDS398 52.36% (Table 2). Our results showed that the mutation efficiency in the LuPDS22 site is 100%, followed by LuPDS398, while the lower mutation efficiency in the other two targets. These results support the previous finding that a GC content of 50%-70% in target sequences usually leads to higher editing efficiency.31,43
However, a previous study reported that the higher GC content may also associated with higher off-targeting. 44 Four potential off-target sites were selected for further analysis, and the sequencing results showed that no mutations were found (Table 3, Supplementary Fig. S1), which indicates that the off-target events of PDS in flax were very few. It is similar to the off-target analysis of other plant species PDS genes in gene editing.19,33,39,45 Taken together, our findings prove that the CRISPR-Cas9 system will be an effective tool for studying gene function in flax.
In recent years, transcriptome sequencing technology in flax has identified genes that have high potential for manipulating fiber quality. 46 For example, 34 LusHSF genes were identified as high-temperature stress responsible genes. Researchers designed gRNAs for individual LusHSFs for gene functional studies via gene editing in future studies. 29 Important functional genes in flax laid a good foundation for the future application of gene editing in improving the quality of flax. In the previous review, researchers have reported that genome-editing strategies can be used to unravel the functions of different members of the tubulin and CesA gene families in flax cell wall biosynthesis, as well as their interactions. 47 Although, they had not discussed the details of genome-editing strategies tailored to specific genes, CRISPR-Cas9 application in flax will contribute to the dissection of roles that specific members of both families. 47 Overall, the CRISPR-Cas9 system is a powerful and precise method to induce targeted mutagenesis in plants.
Conclusions
In summary, the utilization of CRISPR-Cas9 technology to create a flax genome editing system presents exciting opportunities for the genetic manipulation of flax. This research highlights the potential for genome editing to produce superior flax cultivars that are more accessible for cultivation, ultimately benefiting the flax industry.
Footnotes
Acknowledgments
The authors thank Dr. Qijun Chen for providing the CRIPSR-Cas9 genome targeting vector. This work was supported by China Postdoctoral Science Foundation (2022M712192), Shenzhen Commission of Science and Technology Innovation Projects (JCYJ20190808143207457, JCYJ20180305124101630, and JCYJ20170818094958663).
Authors’ Contributions
All authors contributed to the study conception and design. W.Y. and C.W. designed the study. Material preparation, data collection and analysis were performed by C.W., C.S., L.S.: The first draft of the article was written by C.S. and C.W. J.Z., S.L., and Y.B. participated in the statistical analysis and improved the article. All authors commented on previous versions of the article. All authors read and approved the final article.
Disclaimer
All the methods mentioned above were carried out in accordance with relevant guidelines and regulations. The seeds of the flax variety used in this study were obtained from Zhangjiakou Agricultural Science Academy and were allowed to be used in this research.
Ethics Approval and Consent to Participate
This study does not entail any testing on humans or animals. All plant materials utilized in the research adhere to both national and international protocols, as well as local laws and regulations. Furthermore, the use of these plant materials poses no threat to other species in their natural habitats.
Availability of Data and Materials
The Sanger sequencing results from this study have been deposited in the GenBank database (![]() ) with reference accession numbers OQ831697 to OQ831708. The published article and its supplementary information files contain all the data that were generated or analyzed during the study. If needed, the corresponding author can provide the datasets used and/or analyzed during the study upon reasonable request.
) with reference accession numbers OQ831697 to OQ831708. The published article and its supplementary information files contain all the data that were generated or analyzed during the study. If needed, the corresponding author can provide the datasets used and/or analyzed during the study upon reasonable request.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
The work was supported by the Shenzhen Commission of Science and Technology Innovation Projects (JCYJ20190808143207457, JCYJ20180305124101630, JCYJ20170818094958663), Postdoctoral Research Foundation of China (2022M712192), Key Research and Development Program of Shaanxi (2024SF-YBXM-470), General project of Chinese Medicine Administration of Shaanxi Province (SZY-KJCYC-2023-026).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.