
Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by mutations in either the TSC1 or TSC2 genes. Though TSC causes the formation of nonmalignant tumors throughout multiple organs, the most frequent causes of mortality and morbidity are due to neurological complications. In two-thirds of cases, TSC occurs sporadically and TSC2 pathogenic variants are approximately three times more prevalent than TSC1 pathogenic variants. Here, we utilized CRISPR-Cas9-mediated homology directed repair in patient induced pluripotent stem cells (iPSCs) to correct two types of TSC2 pathogenic variants generating two isogenic lines. In one line, we corrected a splice acceptor variant (c.2743-1G>A), which causes the skipping of coding exon 23 and subsequent frameshift and introduction of a stop codon in coding exon 25. In the second line, we corrected a missense variant in coding exon 40 within the GTPase-activating protein domain (c.5228G>A, p.R1743Q). The generation of TSC2 patient iPSCs in parallel with their corresponding CRISPR-corrected isogenic lines will be an important tool for disease modeling applications and for developing therapeutics.
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by mutations in either the TSC1 or TSC2 genes, which affects nearly 1 in every 5,500 newborns, ∼80,000 people in the United States and 2 million people worldwide.1–3 TSC1 and TSC2 encode for Hamartin and Tuberin, respectively, and together with TBC1 domain family member 7 (TBC1D7) form a TSC protein complex which negatively regulates mammalian target of rapamycin complex 1 (mTORC1) signaling.4,5 Dysfunction of the TSC protein complex results in constitutively active mTORC1 signaling, leading to the formation of non-malignant tumors/benign growths throughout multiple organs, predominantly in the brain, kidney, lungs, eyes, and heart.4,6 Though TSC affects multiple organ systems, the most frequent causes of mortality and morbidity are due to neurological complications. Such complications include refractory epilepsy, autism, neuropsychiatric issues, and uncontrolled ependymal growth lesions resulting in life-threatening hydrocephalus.4,7,8 As epilepsy occurs in approximately 85% of patients, the inability to control seizures is one of the most debilitating aspects of the disease as most existing medications show loss of efficacy within 1–2 years of treatment.2,9
Numerous studies have investigated genotype–phenotype correlations in TSC. Although clinical heterogeneity occurs in patients with TSC, one of the most consistent and replicated genotype–phenotype correlations is that patients containing TSC2 pathogenic variants are associated with a more severe clinical outcome than patients with TSC1 pathogenic variants.10–12 This observation is logically consistent with the fact that TSC2, which encodes for a Guanosine-5'-triphosphate (GTP)ase-activating protein (GAP) domain within its C-terminus, is the catalytically active component of the TSC protein complex. Although TSC2 pathogenic mutations occur throughout the gene, several studies have indicated that pathogenic variants cluster within exons 35–40 encoding the TSC2 GAP domain.10,13 This observation is also congruent with another study demonstrating that certain TSC2 variants clustered within exons 23–33 are correlated with a lower risk of infantile spasms. 14 Similarly, variants that reside in the alternatively spliced exons 25 and 31 are highly unlikely to cause TSC. 15
To study the functional significance of pathogenic missense variants in TSC2, induced pluripotent stem cells (iPSCs) provide a powerful in vitro platform since they have the remarkable ability to differentiate into all cell types of the human body including those implicated in neurological manifestations in TSC such as neural progenitor cells (NPCs), neurons, and glia.16–20 Indeed, multiple studies have highlighted the use of directly differentiating patient-specific iPSCs to neural and glial cell types and for generating cerebral organoids (COs), which have provided important mechanistic insights underlying disease pathogenesis in TSC.19,21–26 Since phenotypic variability can arise among healthy control iPSC lines, the generation of corresponding isogenic iPSC lines using CRISPR genome editing tools to correct gene variants remains the gold standard in designing rigorously controlled experiments.27,28
In this study, we generated iPSCs from a patient containing a splice acceptor variant (c.2743-1G>A) in TSC2, which causes the skipping of coding exon 23 and subsequent frameshift and introduction of a stop codon in coding exon 25. In conjunction with a previously generated TSC2 patient iPSC line containing a missense variant in the GAP domain (c.5228G>A, p.R1743Q), we used the CRISPR-Cas9 system to correct both these TSC2 pathogenic variants using homology-directed repair (HDR) methodology resulting in isogenic iPSC lines. Since loss of TSC2 results in mTORC1 hyperactivation, we tested whether astrocytes differentiated from isogenic iPSC lines showed normalization of pS6 levels. In parallel, we also quantified the cell body area of patient and isogenic astrocytes and show a phenotypic rescue of cell body area in both isogenic astrocyte cell lines. Collectively, this data support that gene correction of TSC2 pathogenic variants results in molecular and phenotypic rescue in a clinically relevant cell type of TSC pathogenesis. In addition, the generation of isogenic lines from TSC2 patient iPSCs provide gold standard controls for experimental studies aimed at understanding disease mechanisms and developing therapeutic options for TSC.
Materials and Methods
Generation and maintenance of patient iPSCs
Tuberous Sclerosis Complex Type 2 patient fibroblasts were purchased from the Coriell Institute (GM06100). Using the CytoTune-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific, A16517), patient fibroblasts were induced to pluripotency according to manufacturer’s instructions. Fibroblast reprogramming was performed using Sendai viral vectors driving expression of OCT4, SOX2, KLF4, and c-MYC at a multiplicity of infection of 5. The infected fibroblasts were first cultured in StemPro-34 serum-free medium (SFM) (Thermo Fisher, 10639011) for 2 days and then continued culture in StemFlex medium (Thermo Fisher, A3349401) for approximately 10–14 days. iPSC colonies were first identified using phase-contrast microscopy to identify iPSC colony morphology. These iPSC colonies were picked manually by cutting each colony into small pieces with a P200 pipette tip using a phase-contrast microscope.
In addition, iPSCs from a different TSC2 patient were purchased from the Coriell Institute (GM25318) for CRISPR-Cas9 gene correction. Alkaline phosphatase activity of iPSC lines was determined using the Alkaline Phosphatase Live Stain kit (Thermofisher) according to manufacturer’s instructions. All lines were maintained under feeder-free and defined, serum-free medium conditions. iPSCs were cultured in StemFlex™ Medium (ThermoFisher) and passaged on vitronectin-coated tissue culture plates using standard methods.
Design of sgRNA and homology-directed repair templates
Small-guide RNAs (sgRNA) were designed using Benchling and purchased through Synthego to target intron 24 of TSC2 in patient 6100 (c.2743-1G>A) and coding exon 40 of TSC2 in patient 25318 (c.5228G>A). The sgRNA design was based on the UCSC Genome Brower using the hg38.p10 human genome assembly and RefSeq NM_000548.5 transcript. For each type of TSC2 mutation, a symmetric donor repair template was designed containing the TSC2 wild-type sequence designed with homologous single-stranded (ss) DNA oligonucleotides (Integrated DNA Technologies [IDT]; Alt-R™ HDR Donor Oligos) flanking ∼100 base pairs (bps) upstream and downstream of the cut site. Silent mutations were also introduced within the protospacer adjacent motif (PAM) sites to prevent Cas9 re-cleavage for each TSC2 mutation near the targeted c.2743G>A mutation at c.2746C>A and near the targeted c5528G>A mutation at c.5526G>A. Synthego’s online analysis tool was used to analyze potential off-target sites and both sgRNAs showed no coding genes targeted with fewer than three mismatches. Further, the DNA-PK inhibitor, AZD-7648, was used a final concentration at 10 µM to inhibit nonhomology end joining and boost homology-directed repair, thus further minimizing the potential of incorporating indels into off-target sites.
Nucleofection of CRISPR-Cas9 machinery for gene correction
TSC2 patient iPSCs were nucleofected with a combination of the sgRNA, ssODN, and Cas9 protein using the Neon™ Transfection System. In brief, ribonucleoprotein (RNP) complexes were assembled for each reaction with 2 μL 30 μM sgRNA, 0.5 μL Streptococcus pyogenes Cas9 (SpCas9) nuclease with two nuclear location signals (2NLS), and 3.5 μL of NeonTM Resuspension buffer R. Then the RNPs were incubated at room temperature for 10 min. Prior to nucleofection, iPSCs were incubated for 2 h in StemFlex medium without antibiotics and 10 μM of the ROCK inhibitor, Y-27632. iPSCs were then dissociated with TryPLE (GibcoTM, 12604013) for no >10 min and neutralized with StemFlex medium. 0.25 × 10^6 iPSCs were resuspended in 25 μL of resuspension buffer R (provided with the Neon™ Transfection System 10 μL Kit). Six microliters of RNPs and 1 μL of HDR donor oligos (100 μM, IDT) were combined with 5 μL iPSC suspension as the final transfection mix. The following nucleofection protocol was used with the Neon Electroporation System: 1100 V, 20 ms, and 2 pulses. Nucleofected iPSCs were seeded to a vitronectin-coated 24-well plate with fresh StemFlex media without antibiotics in the presence of the small molecule, AZD7648 (MedChemExpress, HY-111783), at a final concentration of 0.5 µM to enhance homology directed repair. CRISPR-edited cells were then allowed to incubate for 24 h before replacing media with StemFlex media containing antibiotics.
Single-cell clone sorting and genotyping
To screen the pool of CRISPR-edited iPSCs, we generated clonal iPSCs lines by sorting and seeding single iPSCs into vitronectin-coated 96-well plates in Stem Flex media in the presence of a cocktail of small molecules called CEPT 29 (FujiFilm) at 1× final concentration that enhances stem cell survival. The CYTENA UP.SIGHT was used for single-cell dispensing and imaging. Approximately 30 CRISPR-edited iPSC clones were expanded to a 24-well format from single cells from both TSC2 patient iPSC lines. Once clonal iPSCs reached confluency in one well of a 24-well plate, half of the cells were used to extract genomic DNA extraction using the QIAGEN DNeasy Blood &Tissue Kit (QIAGEN, 69504), and the other half of iPSCs were cryopreserved. Polymerase chain reaction (PCR) was performed at gene edited sites using ZymoTaq PreMix and primers shown in Supplementary Table S1 to amplify ∼150 bp upstream and downstream of the mutation site. Sanger sequencing was performed by GENEWIZ™ from Azenta Life Sciences to confirm successful gene correction and introduction of the silent gene edit.
Off-target analysis
Gene-corrected iPSCs were tested for off-target editing events. The most likely off-target sites for sgRNAs used in this study were determined using the Guide Verification Tool by Synthego, which generated a list of off targeted loci containing no fewer than three mismatched nucleotides to our sgRNAs. To evaluate for off-target cleavage, PCR was used to amplify ∼ 100–200 bp upstream and downstream of the predicted off-target cut site (primers shown in Supplementary Table S2). Sanger sequencing was performed on the PCR product by GENEWIZ™ from Azenta Life Sciences.
Chromosome analysis
iPSCs were grown until they reached 40–70% confluency then detached using TrypLE, taking care not to exceed 7 min of exposure to avoid unwanted cell death. After detachment, iPSCs were centrifuged at 300 g for 5 min to form a pellet. The cells were then resuspended in the culture medium containing 0.1 μg/mL Colcemid® and incubated at 37°C for 30 min. The iPSC media was then removed, and the cells were gently resuspended in a hypotonic solution. The cells were incubated at 37°C for another 30 min. Carnoy’s fixative was added slowly with gentle mixing, and the cells were incubated at 4°C for 30 min then further washed with fixative. Cell suspensions were dropped onto slides, dried, then baked at 90°C for 1 hour, followed by overnight desiccation in a vacuum desiccator. Slides were then sequentially dipped into a series of staining dishes: first in a 0.25% trypsin solution in isotonic buffered saline, then in isotonic buffered saline, Gurr buffer with Wright Stain (pH 6.8), and double-distilled demineralized water for a final rinse. Twenty metaphase spreads were analyzed, and karyograms were prepared using MetaSystems software.
Immunofluorescence staining
Cells were grown in a 24-well format and first washed with PBS before being fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, 15713)/DPBS (Gibco, 14190–144) for 15 min at room temperature. PFA was then aspirated and the fixed cells were washed with PBS twice. Following fixation, cells were incubated with blocking solution containing 0.1% Triton X-100/10% donkey serum/Tris buffer solution (TBS) for one hour at room temperature. Cells were then incubated with primary solution (blocking solution, antibodies per dilutions described in (Supplementary Table S3) for 36–48 h at 4°C with consistent gentle shaking. Cells were then rinsed two times with TBST (TBS with 0.1% Tween® 20) solution and then one time with TBS. Then cells were incubated with a blocking solution containing fluorescently-labeled secondary antibodies for 2 h at room temperature. After this incubation period, cells were washed three times with TBS containing 0.1% Triton-X. Nuclei were stained with 1 µg/mL 4′,6-diamidino-2-phenylindole in TBS for 15 min. Images of immunofluorescence staining were captured using a scanning laser confocal microscope (Nikon AX R Confocal Microscope System).
Mycoplasma detection
Mycoplasma contamination was performed using the Mycoplasma PCR Test Kit (Applied Biological Materials) according to the manufacturer’s instructions. The positive control was included in the kit.
STR analysis
Genomic DNA from iPSC lines was extracted and underwent short tandem repeat (STR) profiling analysis to authenticate purity of the cell lines (LabCorp).
iPSC differentiation
To functionally characterize pluripotency of iPSCs, the Human Pluripotent Stem Cell Functional Identification Kit (R&D, SC027B) was used to differentiate iPSCs into all three germ layers according to the manufacturer’s instructions with slight modifications. In brief, base media was prepared by adding the differentiation base media supplement (50×) into Roswell Park Memorial Institute (RPMI) medium (GibcoTM, 11875085). To prepare ectoderm, mesoderm, and endoderm differentiation media, corresponding supplements were then added to the differentiation base media according to the manufacturer’s protocol. We extended the duration of the differentiation period from 2 or 3 days to 5 days for all three germ layers to allow for efficient cellular maturation. After 5 days of differentiation, the cell cultures were fixed and prepared for immunofluorescence staining for cell-type specific markers to characterize each of the three germ layer cell types (Supplementary Table S3). To differentiate iPSCs into astrocytes, we initially generated NPCs using a previously published protocol with minor modifications. 30 The NPCs were then further purified by magnetic-activated cell sorting using anti-PSA-NCAM microbeads (Miltenyi Biotec) according to manufacturer’s instructions. NPCs were then differentiated into astrocytes using a medium consisting of Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12), 1% N2 supplement, 10% BIT supplement (Stem Cell Technologies), and 10% fetal bovine serum, similar to a previous study. 30
Phenotypic rescue and statistical analyses
The relative fluorescence intensity of phospho-S6 ribosomal protein (pS6) and cell size of iPSC-differentiated astrocytes were determined in ImageJ. First, we randomly selected 100 Glial fibrillary acidic protein (GFAP)-positive astrocytes for analysis. For both analyses, lines were manually drawn around the perimeter of each cell. Nuclei were excluded from fluorescence intensity calculations, as pS6 is expressed in the cytoplasm. Statistical analyses were performed in GraphPad Prism v.10.1.0, and data are shown as the mean ± the standard deviation. Statistical significance was analyzed using unpaired T-tests, comparing isogenic control and TSC2 patient-derived astrocytes. Symbols of statistical significance were used as follows: **p ≤ 0.001; ***p ≤ 0.0001.
Results
TSC2 patient iPSC generation and characterization
Fibroblasts from TSC2 patient 1 (6100) containing a splice acceptor variant (c.2743-1G>A) in TSC2 (Supplementary Table S4) were reprogrammed into human iPSCs using Sendai virus-based delivery of Yamanaka transcription factors. Approximately 2 weeks post-infection, reprogrammed fibroblasts were evaluated for characteristic features of human pluripotent stem cells. Emerging iPSC colonies contained tightly packed cells with distinct cell borders and prominent nucleoli, consistent with established iPSC morphology, and alkaline phosphatase activity (Fig. 1). Characteristic stem cell morphology and alkaline phosphatase activity were also confirmed in a previously generated iPSC line (TSC2 Patient 2: 25318) containing a missense variant in the GAP domain (c.5228G>A, p.R1743Q) of TSC2 (Fig. 1; Supplementary Table S4).
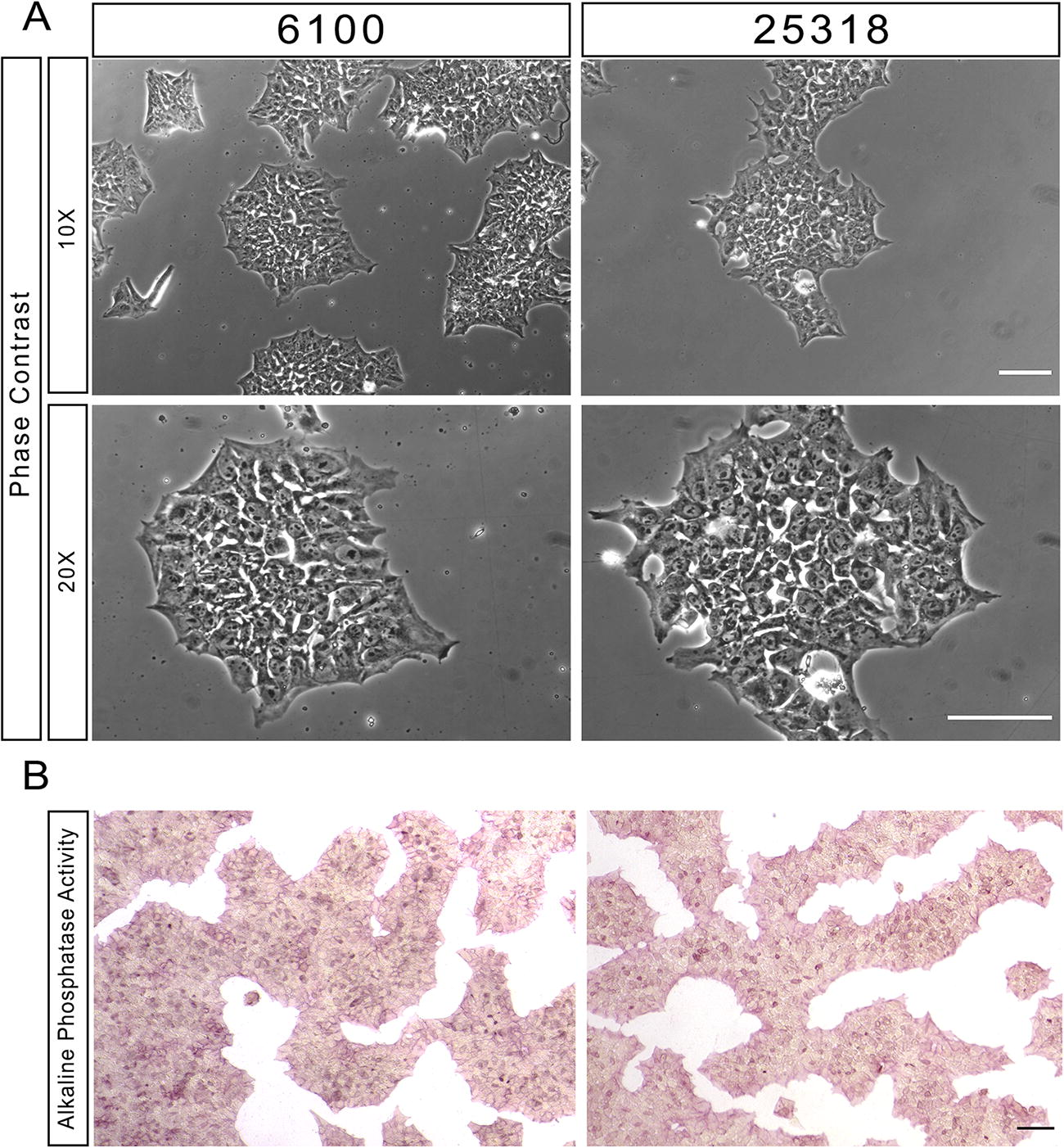
TSC2 patient-derived iPSC lines show alkaline phosphatase activity and morphological hallmarks of pluripotency.
Next, we evaluated well-known pluripotency markers in our iPSCs using immunofluorescence analysis. Both 6100 and 25318 TSC2 patient iPSC lines were positive for all markers tested, which included OCT3, SOX2, NANOG, TRA1-60, TRA1-81, LIN28A, SSEA3, and SSEA4 (Fig. 2; Supplementary Table S3). We also functionally characterized the differentiation potential of TSC2 iPSC lines by testing whether they could differentiate into all three germ layers (Fig. 3A). Both 6100 and 25318 iPSC lines were directly differentiated into all three germ layers using known factors and compounds to promote either ectoderm, mesoderm, or endoderm differentiation. To confirm differentiation status, iPSCs were stained for germ layer-specific markers and both lines showed evidence of ectoderm (Fig. 3B), mesoderm (Fig. 3C), and endoderm (Fig. 3D) differentiation. Lastly, both lines at approximately passage 7 were tested negative for mycoplasma (Supplementary Fig. S1), authenticated using STR analysis (Supplementary Table S5), and 100% of cells showed a normal karyotype in 6100, confirming chromosomal integrity, as performed by G-banding karyotyping (Supplementary Fig. S2). In 25318, 85% of analyzed metaphases showed a normal karyotype and 15% showed an interstitial deletion of chromosome 18q (Supplementary Fig. S2 B,C). Since this iPSC line was procured from the Coriell Repository and was previously karyotyped to be 100% normal, it is possible that this abnormality occurred at a later passage in culture (passage 7). As in this case, routine karyotyping is critical to maintain iPSC cultures with complete chromosomal integrity. Further, biobanking earlier passage iPSCs provides a safeguard if in fact chromosomal abnormalities arise. Collectively, these results authenticate both TSC2 patient iPSCs, which show key molecular, cellular, and functional characteristics of pluripotency in addition to showing cell line purity by STR analysis and a normal karyotype.

TSC2 patient-derived iPSC lines show multiple markers of pluripotency. Immunostaining of iPSC lines for eight different iPSC pluripotency markers. DAPI was used to stain all nuclei. Scale bar = 100 µm. DAPI, 4′,6-diamidino-2-phenylindole; iPSCs, induced pluripotent stem cells.
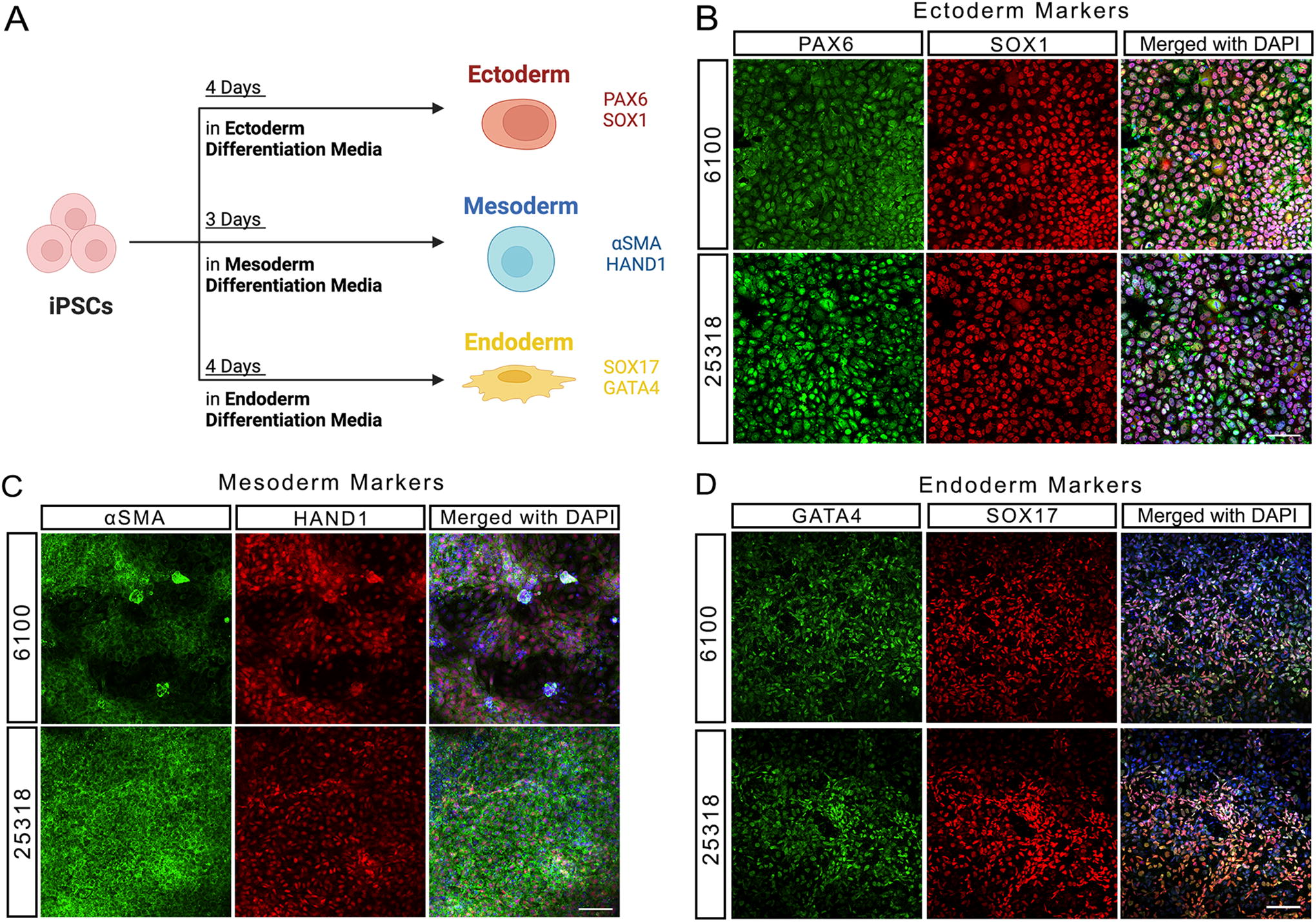
TSC2 patient-derived iPSC lines can differentiate into all three germ layers.
Correction of c.2743-1G>A and c.5228G>A, TSC2 mutations
To generate corresponding isogenic iPSC lines from both TSC2 patient iPSC lines, we used CRISPR-mediated homology-directed repair to perform single bp gene correction either in intron 24 (6100) to correct a splice acceptor site or in coding exon 40 (25318) of TSC2 (Fig. 4). Small guide (sg) RNAs were designed to contain <2 bp matches to other sequences in the genome to ensure precise gene targeting (Supplementary Table S1). To correct the splice acceptor mutation in the 6100 iPSC line, we were fortunate to identify a sgRNA also spanning the mutation site, thereby allowing greater on-target specificity. The cut sites for each Cas9/sgRNA complex were <10 bps away from the mutation sites—9 bp downstream for TSC2 patient 1 (6100) and 3 bp for TSC2 patient 2 (25318) (Fig. 4A and B). We designed ∼100 bp ss DNA donor templates, which provided ∼50 bp homology arms. We also designed silent edits in the PAM sites for each sgRNA to exclude Cas9 from re-cutting the edited DNA site after the initial genetic modification to boost overall gene editing efficiency.
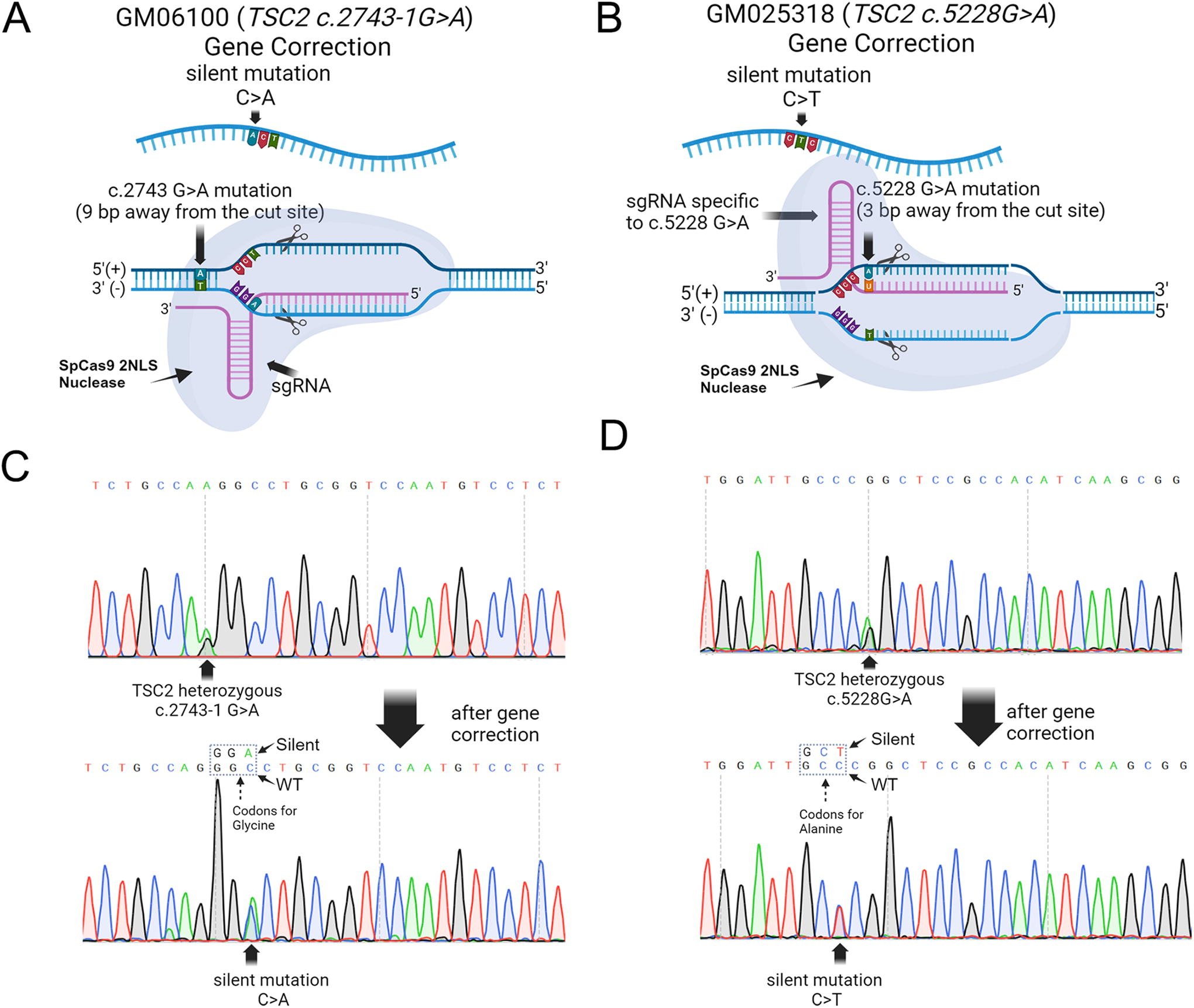
TSC2 mutations in patient-derived iPSCs can be gene corrected using CRISPR-Cas9-mediated homology-directed repair.
The sgRNA was then complexed with Cas9 to generate a RNP complex and together with the single-stranded oligonucleotide donor (Supplementary Table S1) were nucleofected into TSC2 patient iPSCs. To enhance HDR, we incubated nucleofector cells in the small molecule selective DNA-PK inhibitor, AZD-7648, 31 which inhibits nonhomology end joining and thereby promoting HDR. Synthego’s Inference of CRISPR Edits (ICE) software tool was then used to infer the frequency of successful gene editing. We performed PCR amplification of both TSC2 loci that were corrected and then Sanger sequencing to determine correct CRISPR editing. The ABI sequencer data files were then uploaded to the Synthego ICE analysis website (ice.synthego.com). Then the cell pools were sorted into single cells with CYTENA UP.SIGHT single-cell dispenser. Isolated iPSC clones were then screened using Sanger sequencing to confirm successful gene correction and introduction of the silent gene edit (Fig. 4C and D). After screening 26 clones, we identified three correctly gene edited clones from the 25318 iPSC line (11.5% efficiency) and after screening 34 clones from the 6100 iPSC line, we identified two correctly gene edited clones (5.9% efficiency).
Both gene-corrected iPSC lines at passage 3 had normal karyotypes, confirmed through G-banding karyotyping (Supplementary Fig. S3). The corrected iPSCs were then authenticated using STR analysis, revealing an identical profile between the patient- and gene-corrected iPSC lines (Supplementary Table S5). Interestingly, we observed an additional 4 bp repeat at the vWA locus in the 25318 isogenic iPSC line (vWA genotype: 17,18) compared to the patient iPSC line (vWA genotype: 17), which is expected to have no impact on the integrity of this line. The VWA locus is located within intron 40 of the von Willebrand Factor gene and is prone to mutate at a high rate. Additionally, all iPSC lines tested negative for mycoplasma contamination (Supplementary Fig. S1).
Next, we determined the accuracy of our gene correction strategy and tested for the presence of unintended, off-target genome editing. We used Sythego’s Guide Verification Tool, which showed there were no off-target sites with fewer than three mismatches to our TSC2 sgRNAs (Supplementary Table S2). From this list, we focused our analysis on the top four potential off-target sites containing sgRNA sequences with three mismatches to our TSC2 sgRNAs used for gene correction in each gene corrected iPSC line. We PCR amplified these loci and then performed Sanger sequenced and showed there was absence of indels at these loci (data not shown), thus showing the specificity of our CRISPR editing strategy.
Correction of TSC2 mutations results in mTOR pathway normalization and phenotypic rescue in TSC2 patient-astrocytes
Astrocytes play a key role in several aspects of the pathophysiology of TSC, which include decreased calcium entry responding to stimuli, structural abnormalities in mitochondria, and impairment of glutamate buffering. 32 All the pathophysiology phenotypes could potentially contribute to the increased risk of epilepsy. Meanwhile, enlarged astrocytes are often found in TSC patients, which are prone to develop into subependymal giant cell astrocytoma. 33 Therefore, we decided to differentiate the patient-derived and CRISPR-corrected iPSCs to astrocytes to determine whether gene correction could rescue the TSC-related pathological phenotype on both molecular and cellular levels.
We first differentiated patient and corresponding isogenic control iPSC lines into NPCs, followed by magnetic-activated cell sorting purification using anti-PSA-NCAM microbeads, and then differentiated into astrocytes as shown by robust GFAP expression (Fig. 5A). Immunofluorescence staining for phosphorylated S6 ribosomal protein (pS6), a downstream effector of the TSC1/TSC2 complex and mTOR signaling, was performed on astrocytes derived from patient-derived iPSCs and their isogenic controls. Both patient-derived astrocyte lines exhibited elevated pS6 expression compared to their isogenic gene-corrected controls, as expected with mTORC1 hyperactivation. Additionally, the patient-derived astrocytes displayed significantly larger cell bodies relative to the isogenic controls (Fig. 5A–C). This morphological change is consistent with previous reports indicating that mTORC1 hyperactivation leads to enlarged cell body size. Collectively, these results suggest that gene correction effectively restores TSC2 protein function at both the molecular and cellular levels.
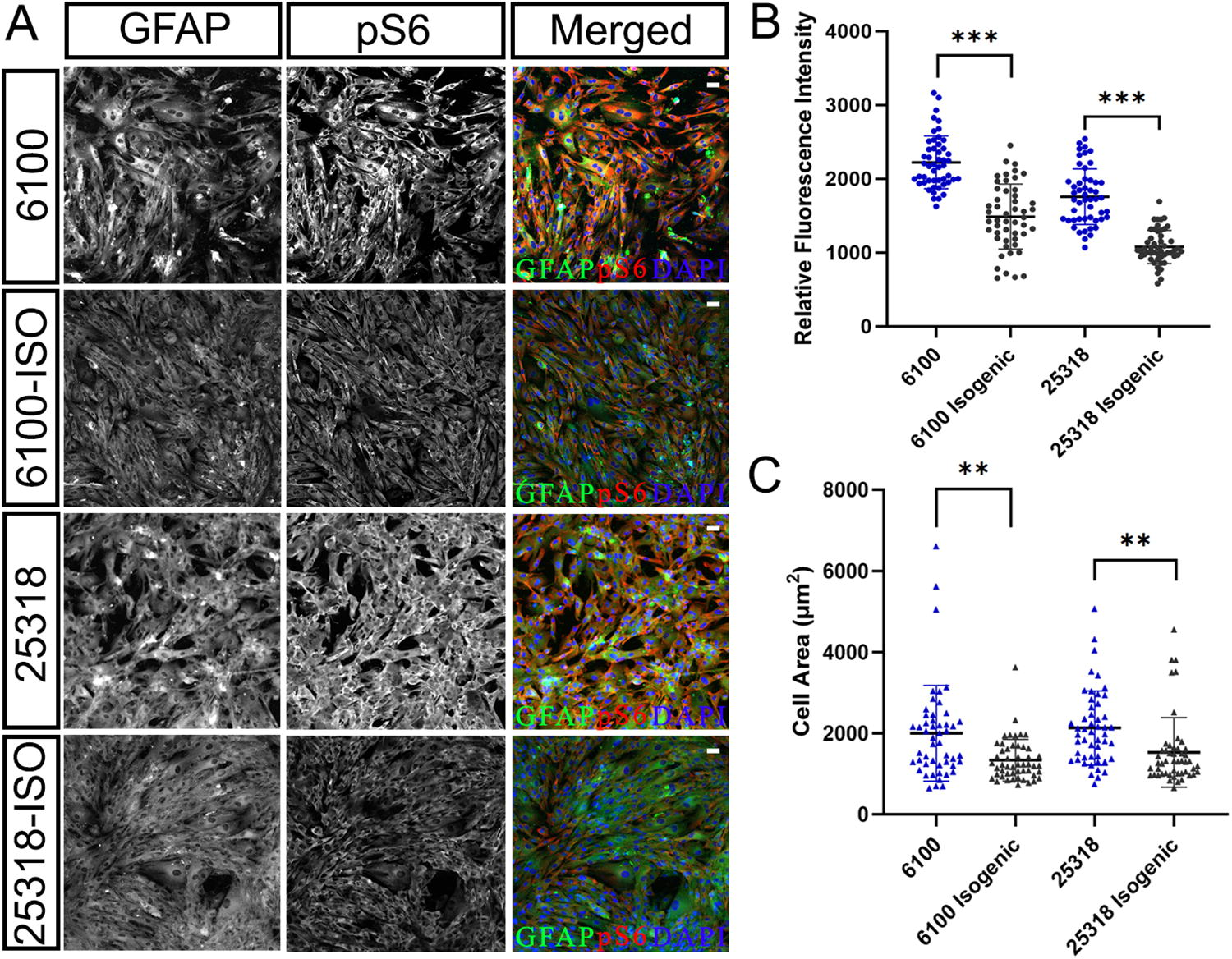
Astrocytes differentiated from gene-corrected lines show molecular and phenotypic rescue of mTORC1 hyperactivation.
Discussion
In this report, we first generated iPSCs from a patient containing a splice acceptor variant (c.2743-1G>A) in TSC2. We also acquired a previously generated TSC2 patient iPSC line containing a missense variant in the GAP domain (c.5228G>A, p.R1743Q) and characterized the morphology and pluripotency of each iPSC line. Utilizing CRISPR-Cas9 editing, we then corrected both these TSC2 variants to generate isogenic iPSC lines. Interestingly, correction of c.5228G>A showed increased CRISPR editing efficiency compared to correction of c.2743-1G>A. It is possible that the sgRNA used to correct c.5228G>A may have been more efficient since it was designed specifically to target the pathogenic mutation, thereby targeting only the mutant allele. Conversely, the sgRNA designed to correct c.2743-1G>A could target both the wild-type and mutant alleles, which could potentially introduce a greater number of indels in the wild-type allele and decrease overall gene correction efficiency at this locus.
Next, we differentiated isogenic and patient iPSCs into astrocytes and assessed whether they showed molecular and phenotypic rescue of mTORC1 hyperactivation. Isogenic and patient iPSCs were differentiated into astrocytes and assessed for molecular and phenotypic rescue of mTORC1 hyperactivation. Both isogenic lines showed a reduction in pS6 levels and cell body size compared to patient lines, suggesting that correction of the TSC2 mutations rescues TSC pathogenesis. These data represent important proof-of-concept in generating gene-corrected, patient-derived iPSCs for human disease modeling and treatment of TSC Type 2.
Studies have only recently begun to utilize patient-derived iPSCs to investigate how mutations in TSC2 impact human neural development and function. Eichmüller et al. utilized iPSCs to develop TSC patient-specific COs, which manifested several clinical pathophysiological features, including proliferative, tumorigenic zones, and dysplastic cortical lesions containing giant cells. 26 Abnormalities have been reported in iPSC-derived neurons and NPCs from TSC patients, including mTORC1 hyperactivation, 34 network hyperexcitability22,23 increased soma size,23,35 increased neurite length, 23 and delayed differentiation. 36 Cerebellar Purkinje cells derived from TSC2 patient iPSCs also displayed mTORC1 hyperactivation, hypoexcitability and reduced synaptic activity, and defects in RNA regulation and neuronal differentiation. 37 The vast majority of TSC patient-derived iPSC studies have focused on NPCs and neurons, with only two recent studies focusing on glia.22,23,35 Hyperactivation of mTORC1, through the loss of TSC1 or TSC2, can lead to glial abnormalities, including astrogliosis, microglial activation, and a decrease in oligodendrocytes.38–40 These glial defects may affect neurogenesis, neuronal function, and lead to an increase in inflammation caused by microglial activation. 41 It has been difficult to isolate the role of glia in neurological manifestations and vice versa, but utilizing patient and isogenic derived iPSCs provides an excellent and simplified tool for further investigation.
For our CRISPR editing strategy, we utilized Alt-R HDR ss DNA oligo donors, which have been end protected using a proprietary modification strategy to increase stability and enhance overall CRISPR editing efficiency. Using this strategy, we achieved 5.9% and 11.5% gene correction efficiency in the 6100 and 25318 iPSC lines, respectively. In a previous study, an improved method to increase gene insertion efficiency was established by chemically modifying 12–19% of internal bases, which was named enhanced ssDNA (esDNA). 42 This strategy significantly increased gene insertion efficiency at the CFTR locus in airway basal stem cells, CD34+ hematopoietic cells (CD34+ cells), T-cells, and endothelial cells compared to end-modified or unmodified templates. However, in iPSCs, there was no advantage of using either esDNA or end-modified templates compared to using an unmodified donor template. It was hypothesized that this was because the 3′ repair exonuclease 1, TREX1, is not expressed in iPSCs. In future studies, it may not be advantageous to use chemically modified donor templates for CRISPR-mediated gene correction strategies in iPSCs.
Conclusion
In this study, we corrected two types of variants in the TSC2 gene, a splice acceptor site in intron 24 (c.2743-1G>A) and a missense variant in coding exon 40 (c.5228G>A, p.R1743Q), in two independent patient-derived iPSC lines. In TSC Type 2 patients, these mutations lead to significant neurological morbidity such as growth of subependymal nodules, refractory epilepsy, and cognitive impairment. We show proof of concept that CRISPR-based HDR using a RNP complex of a sgRNA with Cas9 and a ssDNA donor template is efficient to correct these variants in the presence of the DNA-PK inhibitor, AZD-7648. We further demonstrate that correction of these pathogenic variants normalized mTORC1 levels and cell body size in iPSC-derived astrocytes. Thus, disease models such as the use of COs can be generated using patient iPSC lines in conjunction with these isogenic control lines to dissect molecular and circuit level disease mechanisms underlying TSC. Further, these isogenic iPSC lines will provide important controls for screening novel classes of compounds and for developing novel gene therapy approaches to treat TSC.
Footnotes
Acknowledgments
The authors are grateful to all Hester laboratory members for their advice and constructive critiques related to this study. Figures 3 and ![]() contain schematics that were created with BioRender.com.
contain schematics that were created with BioRender.com.
Authors’ Contributions
G.G., M.M., L.C., D.J., and M.E.H. designed and planned the experiments. G.G., M.M., L.C., D.J., and S.T. carried out and performed the experiments. G.G., M.M., M.E.H., S.T., and C.C. analyzed data and contributed to the interpretation of the results. G.G., M.M, S.T., M.E.H., P.R., and C.C., wrote and edited the article. All authors provided critical feedback and contributed to the overall article.
Author Disclosure Statement
The authors report no competing interests.
Funding Information
The funding of this work was generously supported by the
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.