
Abstract
Sickle cell disease (SCD) is a hereditary blood disorder caused by a specific mutation in the β-globin gene, leading to the production of hemoglobin S, which deforms red blood cells, causing occlusion in small blood vessels. This results in pain, anemia, organ damage, infections, and increased stroke risk. Treatment options, including disease-modifying therapies and curative hematopoietic stem cell transplants, have limited accessibility. Recently, autologous gene therapy has emerged as a promising curative option, particularly for SCD. Gene editing techniques such as CRISPR, base editing, and prime editing offer potential to correct this mutation. In this review, we discuss recent preclinical studies and clinical trials of gene and cell therapies, focusing on the progress of FDA-approved treatments like Lyfgenia and Casgevy. We also examine the many challenges, including accessibility, safety, and long-term efficacy, which continue to shape the future of SCD gene therapy.
Introduction
Millions worldwide are affected by sickle cell disease (SCD), 1 a hereditary blood disorder affecting individuals of African, Mediterranean, Middle Eastern, and South Asian descent. 2 The first molecular disease, as Linus Pauling called it, SCD is caused by a mutation in the β-globin gene (HBB) resulting in the production of hemoglobin S (HbS). This is an abnormal hemoglobin that affects the structure of red blood cells under deoxygenated conditions, resulting in the deformation of red blood cells, which in turn leads to occlusion in the small blood vessels. 3 Downstream effects include severe pain episodes, anemia from destruction of these deformed red blood cells, end organ damage from vaso-occlusion, increased susceptibility to infections, and a risk of stroke. 4 These complications lead to early morbidity and mortality in patients with SCD. In the United States, the median lifespan for individuals with SCD is 42 years for females and 38 years for males. 2
Until recently, there were only a handful of treatment options for SCD available (Fig. 1), including disease-modifying therapies and curative options.
5
Disease-modifying therapies include medications such as hydroxyurea,
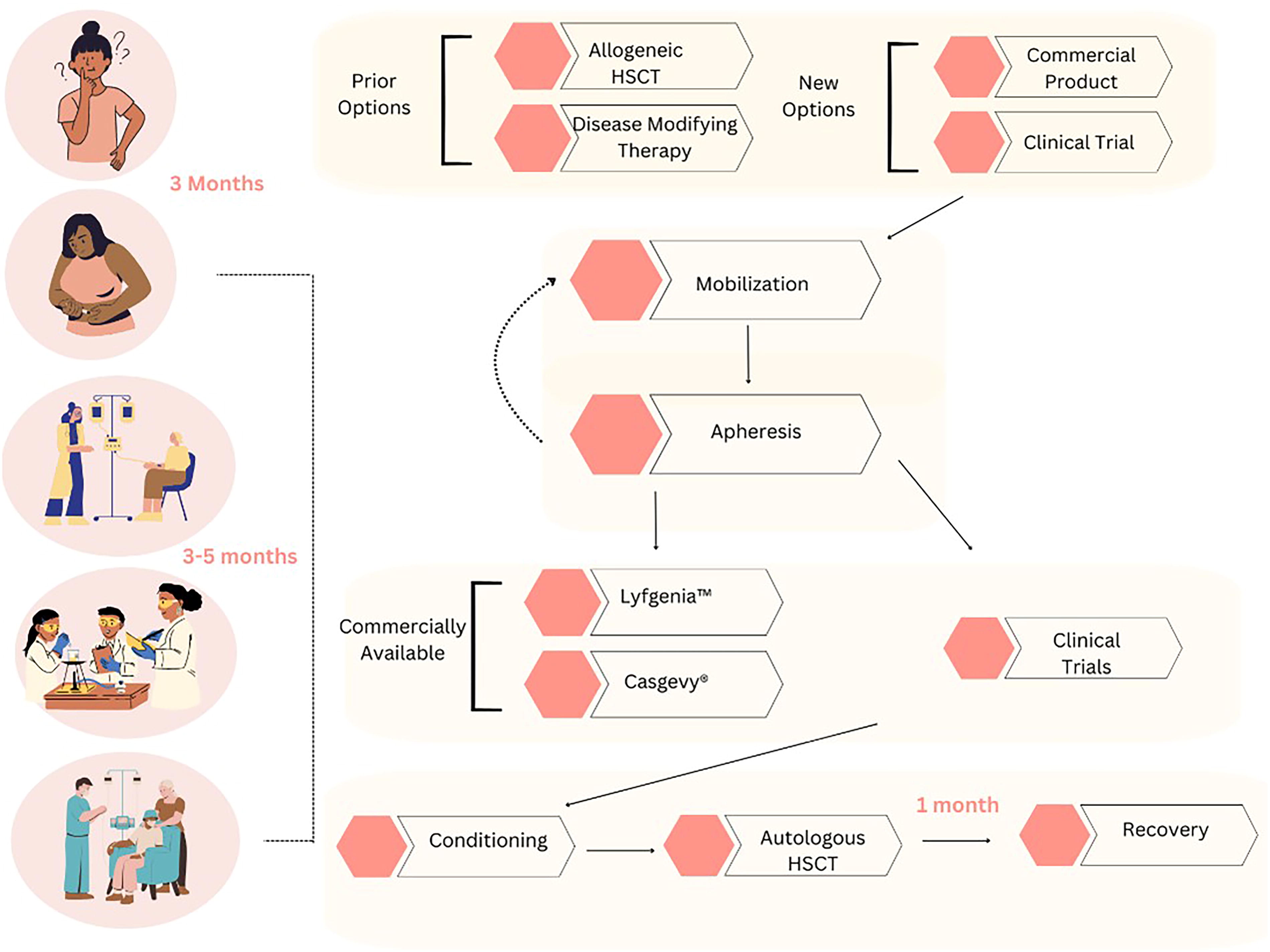
Current treatment options, processes and timeline available to patients with SCD from beginning to end. SCD, sickle cell disease
For these reasons and due to the continual advances in technology, autologous gene therapy has come to the forefront of curative options for sickle cell patients. SCD results from a single point mutation in the HBB gene, which causes a substitution from glutamic acid (E) to valine (V) at the sixth amino acid (E6V), resulting in HbS instead of the wild-type Hb (HbA). 8 As SCD is a monogenic disease, autologous gene therapy is a viable option to treat this condition. There are several different editing techniques being explored, some of which have been successfully delivered to patients, while others remain in development.7,9–11 These techniques include the correction of the underlying mutation in the HBB gene, induction of fetal hemoglobin (HbF) to rescue patients from the sickling phenotype, and the addition of a functional anti-sickling β-globin gene. 12
In this review, we shall focus on gene therapy for patients with SCD from preclinical studies to clinical trials and the recent Food and Drug Administration (FDA) approval (in December 2023) of two landmark therapies for SCD. We will also review access to care and hurdles associated with the delivery of these products to patients.
Gene Editing Techniques
The desired outcome in gene editing is to take a DNA sequence and convert it into a new desired sequence. The selection of the method used will depend on the desired edit. 13 In SCD, several targets have been explored and found to be advantageous. The first is the HBB gene itself, while another commonly studied target is BCL11A, a repressor of γ-globin expression. 14 These genes can be edited to correct the mutation using techniques such as Clustered Regularly Interspaced Short Palindromic Repeats-Cas9 (CRISPR-Cas9), base editing, and prime editing. In this review, we will focus on the techniques that have been applied clinically.
Prior to the emergence of CRISPR-Cas technology, zinc finger nucleases (ZFNs) were used to study SCD. 15 ZFNs link the DNA-binding domain of zinc-finger proteins (ZFPs) with the nuclease domain of the FokI restriction enzyme. 16 ZFPs consist of a tandem array of Cys2-His2 finger motifs, enabling ZFNs to bind to short DNA sequences. Each ZFP domain targets a specific DNA triplet, and ZFNs work as a pair of nucleases that recognize sequences on both the forward and reverse DNA strands surrounding the target site, binding to each side. Once bound, the FokI domain pair induces a double-stranded break (DSB) at the target site, cleaving the DNA. 17
However, ZFNs have limitations in targeting specific DNA sequences, and the lack of specificity in some zinc-finger domains can result in off-target cleavage. Moreover, using ZFNs to target precise DNA sequences is a complex and labor-intensive process.18,19 This has led to a focus on CRISPR and other more accessible techniques. Bacteria use RNA-guided endonucleases of CRISPR-Cas systems to recognize and cleave foreign nucleic acids. They retain a record of previously encountered pathogens by capturing nucleic acid sequences known as “spacer sequences” for future encounters. 20 Using different spacer sequences within a guide RNA, we can reprogram these systems to target DNA and RNA sequences as long as the matching target DNA protospacer sequence is located next to a suitable protospacer-adjacent motif (PAM). 21 Cas9 effectors are RNA-guided endonucleases that generate DSBs in target DNA sequences. 22 These breaks are then repaired by different DNA repair machinery that are endogenous to cells.
Another form of editing involves the addition of a functional gene to compensate for the loss of a functional β-globin gene. This includes the addition of functional genes that can be delivered to the genome by means of a viral vector. 23 There are several viral vectors that can be used such as lentivirus and adeno-associated virus (AAV). This technique, known as transduction, has wide applicability for cell types, high transfection rate, minimal immunogenicity, and minimal cytotoxicity. 24 However, there are packaging restrictions depending on the vector selected. 25
Building on the CRISPR platform, base editors have emerged as a very promising technology to target and modify single-base mutations. They do not require the formation of DSBs or donor DNA template as is required using CRISPR. 13 As a result, fewer deleterious insertions and deletions are created. Base editing involves a nonfunctioning CRISPR-Cas nuclease combined with a single-stranded DNA deaminase. Base editors can efficiently mediate all four possible transition mutations (C→T, A→G, T→C, G→A), which represent approximately 30% of currently annotated human pathogenic variants.
Prime editing is another precision genome editing technique that is being studied in the context of SCD correction.26–28 The prime editor protein is a combination of Cas9 and a highly engineered reverse transcriptase (RT), which works together with a prime editing guide RNA (pegRNA). The pegRNA targets the specific editing site and encodes the necessary edit. Prime editing enables precise insertions, deletions, and all 12 types of point mutations with minimal off-target effects, as the correction is directly transcribed from the pegRNA by RT. 13
While gene editing techniques offer groundbreaking potential for treating SCD, their efficacy and safety have been rigorously tested through pivotal preclinical studies that have laid the foundation for clinical application.
Pivotal Preclinical Studies
Various therapeutic strategies are being investigated for treating SCD, including gene addition, gene editing, and gene regulation techniques. 12 The path to FDA approval for gene therapies targeting SCD has been marked by groundbreaking preclinical trials that established the safety and efficacy of these treatments. Here, we will focus on the pivotal preclinical studies related to autologous HSCT.
Lyfgenia is a gene therapy developed by Bluebird Bio that involves the use of a lentiviral vector to insert a functional copy of the β-globin gene into a patient’s hematopoietic stem cells. 29 The goal is to compensate for the underlying genetic defect by means of gene addition. Multiple preclinical studies have reported the approach using the βT87Q-globin gene to reduce the production of HbS in red blood cells. It was tested in differentiated human erythroid cells ex vivo and demonstrated a significant reduction in HbS levels and improved anti-sickling properties. 30 In 2001, Leboulch et al. supplied in vivo data in genetically modified mice that model the human condition. 31 By employing a gene therapy approach that involves introducing a functional copy of the β-globin gene into mouse hematopoietic stem cells, researchers achieved significant restoration of normal hemoglobin production and a reduction in HbS levels. The treated mice showed improved red blood cell function and reduced disease symptoms, demonstrating that the gene therapy effectively addressed the genetic defect underlying SCD. These findings suggested that βT87Q-globin gene therapy could effectively mitigate SCD by decreasing HbS production and enhancing red blood cell function, paving the way for clinical applications.
Gene editing technologies, particularly CRISPR-Cas9, have also played a pivotal role in developing new therapies for SCD, with particular emphasis on editing the BCL11A repressor. This approach led to clinical trials (sponsored by Vertex Pharmaceuticals and CRISPR therapeutics) for Casgevy, the first CRISPR-Cas9 therapy available to patients with SCD. This gene editing technique focuses on disrupting the BCL11A repressor of HbF production, which is normally shut down in the months after birth.23,33 Increasing HbF levels can alleviate SCD symptoms due to its anti-sickling properties, as evidenced by SCD patients who also carry mutations leading to HPFH, resulting in fewer or no symptoms.11,34–36
Researchers have explored two pathways to upregulate HbF: a BCL11A knockout or targeting the BCL11A enhancer. 37 Direct knockout of the BCL11A gene leads to the complete loss of its function, ensuring a strong effect in terms of gene silencing. However, this may inadvertently disrupt nearby genes leading to off-target effects. Further, BCL11A is involved in other tissues, and its complete knockout could lead to adverse effects in nonerythroid cells, such as in the brain or other organs, where the gene appears to play a key role in development. Targeting the erythroid enhancer, which is specifically active in red blood cell precursors, should avoid off-target effects in nonerythroid cells38,39 and offer a safer option.40,41 However, targeting the enhancer might not completely shut down BCL11A, especially in cases where other compensatory mechanisms or enhancer elements exist that could mitigate the effect.
Ex vivo studies showed that genetic modification of human HSCs by BCL11A knockout led to increased HbF production and a reduction in sickling.42–45 Researchers conducted studies using mouse models to investigate the effects of BCL11A knockout and showed increased expression of HbF. 46 Finally, to evaluate safety, other investigators evaluated the off-target effects of BCL11A editing using CRISPR. 47 Recent studies looking at dual editing of the BCL11A enhancer are showing promising results with higher levels of HbF induction. 40 This work has helped refine the CRISPR-Cas9 techniques to minimize risks and maximize the therapeutic potential, thus addressing concerns that could impact the transition to clinical trials.
While gene modulation techniques show promise, they do not address the underlying mutation of SCD. This can lead to ongoing hemolysis, as patients have been shown to produce 45% HbS, which has the potential for complications.48,49 Correcting the sickle cell mutation directly within the patient’s own HSCs to produce normal hemoglobin has also been attempted, though it presents greater challenges than disruption methods.50,51 Preclinical studies have shown promise for this technique, but the first trial testing this approach (sponsored by Graphite Bio) was halted due to an adverse event in the initial patient. 52 This patient has since been followed by Kamau Therapeutics and found to have an initial increase in HbA followed by a downtrend around 7 months with a corresponding increase in HbF <78%. The mechanism of HbF upregulation is currently under investigation. 53
Newer techniques such as base and prime editing have also been studied in preclinical studies. Base editing cannot reverse the SCD point mutation to the wild-type sequence, but the use of an adenine base editor (ABE) produces an E6A genotype, known as Makassar beta-globin (HbG). 54 This is a rare naturally occurring variant that is considered nonpathogenic.55,56 Studies have shown that ABEs can convert HbS to HbG in HEK293T cells with up to 55% efficiency. 57 Similarly, researchers using mouse models have achieved 80% editing from HbS to HbG using ex vivo delivery of ABEs into HSPCs derived from SCD patients. 58 Although Makassar β-globin holds promise as a potential therapeutic approach, further research in clinical trials is needed. Beam Therapeutics has chosen not to move this strategy into the clinic. 59 (Currently, there is no A-to-T base editor available for testing in SCD correction models.)
While base editing and prime editing are both emerging techniques, it is important to recognize when one might be more indicated than the other. Both techniques have their pros and cons. 13 In some preclinical studies, base editors have been shown to have higher editing efficiencies in comparison to prime editors. Nonetheless, preclinical studies have demonstrated above 40% efficiency using prime editing, and in comparison to Cas9-HDR for HBB, some have shown 270-fold higher editing-to-indel ratio. 28
The success of these preclinical studies was pivotal in advancing both Lyfgenia and Casgevy to clinical trials. Both therapies received FDA approval in December 2023 for the treatment of SCD.10,60 The positive outcomes in other preclinical models provided the necessary evidence of safety and efficacy required for initiating human trials that are now ongoing for the treatment of SCD.
Clinical Trials
In Tables 1 and 2, we list the gene therapy clinical trials for SCD based on two approaches: gene addition (Table 1) and gene editing (Table 2). Some trials on long-term effects are listed in Table 3. Here, we focus on trials that have resulted in FDA-approved treatments for SCD.
Gene addition clinical trials
SCD, Sickle cell disease.
Gene editing clinical trials
Source: clinicaltrials.gov.
Long-term follow-up trials
Following promising preclinical studies, Bluebird Bio conducted a single-center study (NCT02151526) evaluating the safety and efficacy of LentiGlobin, an autologous product transfected with the BB305 lentiviral vector Drug Product for patients with β-thalassemia or SCD. This trial included three patients (13, 16, and 21 years old) with severe SCD who safely received LentiGlobin. A follow-up after 5½ years continued to show sustained production of therapeutic HbAT87Q, along with a reduction of vaso-occlusive events and hemolysis. 49 These patients are now under long-term observation in a separate study (NCT04628585).
Based on these promising results, a multicenter study (NCT02140554) phase I/II, open label, nonrandomized study for patients with severe SCD was conducted in three cohorts. Cohort A included seven patients who received LentiGlobin manufactured from bone marrow-harvested CD34+ cells, which was durable but had a lower vector copy number and suboptimal expression of HbA. Significant changes were made for patients in group B, which included transfusions prior to collection to reduce stress erythropoiesis, a higher busulfan dose AUC (>5,000 µmol × min), and a refined manufacturing process to improve transduction efficacy. Group B consisted of two subgroups: Group B1 (n = 1) received total two products, one from previous manufacturing and one from refined process, and Group B2 (n = 1), who received products only from the refined process. These changes improved the quality and quantity of the collected cells as demonstrated by increased vector copy numbers and HbAT87Q production, higher hemoglobin levels, low HbS percentage, and decreased hemolysis. 49
The trial was briefly put on hold in 2021 following a report of two cases from Cohort A with acute myeloid leukemia (AML). Both patients received the product from bone marrow harvests and underwent the early manufacturing process. The first patient was diagnosed with myelodysplastic syndrome, which progressed to AML and death. Investigators determined it was unlikely from the drug product following absence of vector in blast population and attributed secondary to myeloablative chemotherapy. In the second case, vector was present in blast cells in the VAMP4 gene, which is not known to play a role in development of AML and considered unlikely to be a direct result of LentiGlobin.
The trial resumed with 35 patients in cohort C, which received LentiGlobin following further process optimization. This showed sustained and durable engraftment in all 35 patients with sustained pan-cellular expression of HbAT87Q contributing to at least 40% of total hemoglobin and reduced to normalization of hemolysis markers. Two patients with two-gene deletion α-thalassemia trait treated showed persistent anemia with one adult requiring RBC transfusions. The hematological abnormalities were restricted to the erythroid lineage. 49 Among patients evaluated for transplant population with vaso-occlusive events (n = 25), all had resolution of severe vaso-occlusive events.
Vertex Pharmaceuticals and CRISPR Therapeutics conducted a multisite trial, CLIMB-SCD-121 using ex vivo CRISPR–Cas9 technology of gene editing autologous CD34+ HSPCs at the erythroid-specific enhancer region of BCL11A, leading to FDA approval of Casgevy. A total of 44 patients were enrolled in the CTX001 trial; most adverse events were noted secondary to chemotherapy, none from the infusion itself. The median of neutrophil engraftment was 27 days and 35 days for platelets. One death was reported due to SARS-CoV2 infection. In interim analysis, 30 patients met the primary efficacy endpoint and 29 (97%) were free of VOC for about 12 months. In the full analysis, of 43 patients, 37 (86%) were free from severe VOC for up to 45.5 months. There was a sustained increase in hemoglobin levels (12.5+/−1.8 g/dL) and fetal hemoglobin (43.9+/−8.6%) at month 6, and normal or near-normal levels at follow-up and with a mean HbF level of over 40% at follow-up. The mean percentage of F cells (the percentage of red cells expressing HbF) was greater than 90% at follow-up, with improvements of hemolysis markers. About 15 patients had a single α-globin gene deletion, two patients with two α-globin gene deletion; all of them showed similar increased total Hb and HbF levels in comparison with patients who did not have the α-globin gene deletion. The patients in this study will be followed in CLIMB-131 (NCT04208529) for long-term effects.
Although only Casgevy and Lyfgenia have received FDA approval so far, several other clinical trials are ongoing with promising data. Esrick et al.at Boston Children’s Hospital are conducting an ongoing study (NCT03282656) demonstrating a favorable risk-benefit profile in six patients with severe SCD, with a median follow-up of 18 months. Their gene editing approach uses short hairpin RNA (shRNA) targeting BCL11A mRNA embedded in a microRNA (shmiR) via integration with a lentiviral vector. This successfully overcame BCL11A knockdown in other lineages by reducing erythroid-specific lineage cells by 90%. The median time for engraftment was 22 days for neutrophils and 33 days for platelets. Participants experienced substantial and stable increases in HbF levels (20–41% of total hemoglobin), with HbF broadly distributed across red blood cells (59–93.6% of cells contained HbF) and sustained throughout the study. None of the patients experienced VOC, acute chest syndrome, or stroke after treatment. Adverse events were primarily related to the chemotherapy conditioning regimen. While no insertional oncogenesis was noted, lentiviral therapies historically carry theoretical risks, as the BCH-BB694 vector requires genomic integration. Three patients who previously required regular blood transfusions for secondary stroke prevention were able to stop or reduce their transfusion regimen. 61
Renizgamglogene autogedtemcel (reni-cel) is an investigational gene-edited product that uses the Cas12a nuclease to edit the promotor of the γ-globin genes, HBG1/HBG2 (−118 to −113 CCAAT box). This approach mimics mutations in patients with Hereditary Persistence of Fetal Hemoglobin and reactivates γ-globin expression. Preclinical studies have shown ≥80% editing efficiency in CD34+ cells with high specificity and no off-target editing. 62 The RUBY trial (NCT04853576; sponsored by Editas Medicine) demonstrated early, durable correction of anemia with sustained HbF induction (≥40% total Hb with pancellular distribution) and normalization of hemolytic markers by month 6 in 28 patients with SCD, with a median follow-up of 9.5 months. Median engraftment times were 23 days for neutrophils and 25 days for platelets. By month 6, the mean percentage of F cells increased by 99.3%, with levels maintained above 97% throughout follow-up. Of 28 treated patients, 27 remained VOE-free post-infusion as of October 2024. The study also showed early and sustained meaningful improvements in pain, physical, and social patient-reported outcomes. 63
To understand the benefits of HbF induction via BCL11A inhibition compared with hydroxyurea, De Souza et al. used single-cell assays to show that BCH-BB694-treated patients had higher HbF percentages (27.9% vs. 27.0%) and fewer sickle-prone RBCs, defined as RBCs with HbS >70% (42% vs. 61%), compared with high responders to hydroxyurea.64,65
LentiGlobin’s gene addition approach offers a direct replacement of defective hemoglobin, contrasting with the HbF-focused strategies of exa-cel, reni-cel, and BCH-BB694. While effective in reducing SCD complications, its reliance on lentiviral integration introduces safety considerations absent in CRISPR-based therapies. Exa-cel and reni-cel provide durable, precise DNA edits without viral vectors, positioning them as potentially safer long-term options despite similar efficacy profiles. CRISPR-based therapies induce permanent DNA edits, offering a likely advantage in long-term durability, while the long-term durability of shRNA-mediated BCL11A suppression for elevating HbF remains unknown.
Discussion
It has been 18 months since the landmark approval of two gene therapy products—Casgevy and Lyfgenia—for SCD. We are learning important principles and lessons that should guide us forward.
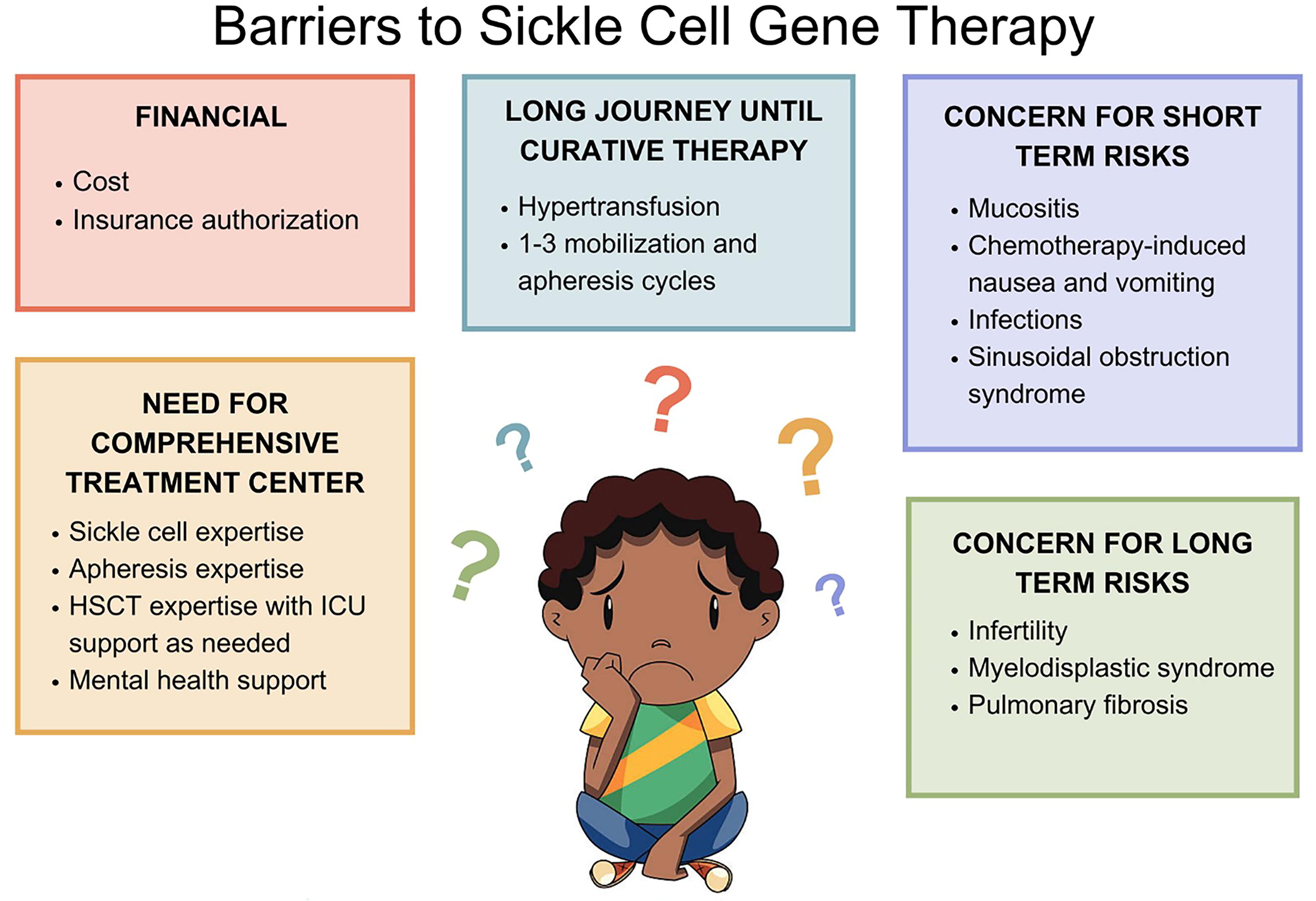
While we anticipate that it will get easier, it remains expensive, difficult, and time-consuming to progress from initial patient consult to curative therapy. Authorization of these products can take many weeks and frequently requires a single case agreement with the payor. Research, regulatory, and manufacturing costs have led to biotech companies setting prices of more than $2 million per product. Before scheduling patients for stem cell collection, authorization must be in place. On a positive note, as of December 2024, both manufacturers (Vertex and Bluebird) had entered into agreements with the Centers for Medicare & Medicaid Services to participate in the Cell and Gene Therapy Access Model. States participating in outcomes-based models for their patients with Medicaid will likely be able to lower the overall cost of delivery of gene therapy. 66
In addition, patients with SCD frequently require more than one stem cell collection. One reason is that filgrastim mobilization cannot be used given the risk of VOC and mobilization with plerixafor alone is not always as effective. 67 Second, even with an adequate stem cell dose, there is often insufficient production of the genetically manufactured product, requiring additional collections. For example, there can be significant loss of stem cells during the electroporation process (used in Casgevy).
Once we send a product for manufacturing with an adequate cell dose, there are several possible outcomes: (1) the center receives genetically modified product that meets cell dose requiring no additional collections; (2) the center receives genetically modified product that does not meet the required cell dose, necessitating additional collection; and (3) genetic manipulation was not successful or the product is deemed out of specification, requiring full recollection. It can take 3–5 months to progress from the last collection to infusion (Fig. 1). During that time, the patient may continue experiencing symptoms such as VOC and acute chest syndrome. Given this complex patient journey 68 requiring insurance approval, multiple stem cell collections, and centers navigating coordination with apheresis, clinical teams, interventional radiology, flow lab, cell therapy lab, and the sponsor, it is no surprise why fewer treatments have been performed to date than was anticipated and hoped for by the companies and the SCD community.
The overall treatment, including transfusions, mobilization, sickle cell care, pain management, mental health support, and delivery of high-dose chemotherapy must occur at a comprehensive medical center capable of handling and coordinating all aspects of patient care. Our patients will not be served well by an excellent sickle cell program without an experienced transplant program and vice versa. The patients coming to gene therapy are some of the most complex sickle cell patients and require very specialized multidisciplinary care. In terms of their myeloablative chemotherapy, using high doses of busulfan, the risks not only of mucositis and nausea/vomiting are high; in addition, the risk of sinusoidal obstruction syndrome is significant enough requiring comfort in the initiation of defibrotide and the use of diuretic drips and/or dialysis as needed. 69
Although we are still in the infancy of this field, if results continue to be successful, uptake of gene therapy will increase as patients, payors, and centers become more comfortable offering this therapy. For now, it is offered as an alternative to haploidentical transplant in eligible patients over 12 years of age, but one may imagine that given no risk of GVHD and lower risks of infection that gene therapy may eventually continue gaining ground.
Alternative donor transplants have become widely accessible (90% donor availability) with impressive outcomes (88% event-free survival, 95% overall survival at two years). 70 They remain the standard of care globally, especially where gene therapy is cost-prohibitive. Transplants work effectively even for patients with organ damage through reduced-intensity protocols.
Gene therapy, while eliminating donor needs and GVHD risks, costs 5–6 times more than transplantation ($2–3 million vs. $467,747) and often excludes patients with organ damage due to intensive chemotherapy requirements. Despite current limitations, gene therapy could revolutionize global treatment if made more affordable and accessible. Both approaches offer transformative potential, with treatment selection depending on individual patient factors including donor availability, existing organ damage, and financial considerations.
Over the next 5–10 years, we envision certain aspects of the therapy and its delivery to change, allowing for better access and more patients treated. We expect studies to show the treatment is beneficial in patients younger than 12 as well as in patients with stroke or cerebrovascular events. This will open the door to many new patients. As we have more patients willing to go through the treatment, the companies will increase the manufacturing slots. Both pharmaceutical companies and academia need to focus on certain issues to broaden applicability. Biopharma companies must continue working on their manufacturing process with a goal of reducing cell loss and reducing out-of-specification products. This will hopefully result in many more patients having successful products after a single collection. Academic centers should focus on trials with less-toxic chemotherapy (such as treosulfan, 71 which was recently approved by the FDA), which will lead to milder short term side effects and possibly less infertility. Additionally, if studies with the stem cell antibody CD117 72 show success, this would revolutionize the field as patients are vocal about their concerns of side effects with myeloablative chemotherapy, particularly infertility.
Over the next decade, however, we envision the field shifting to in vivo gene therapy. There are promising preclinical studies looking at the delivery of genetic material using AAV vectors or liquid nanoparticles. 73 The approach may be similar to one used by neurologists with Elevidys used to treat Duchenne muscular dystrophy. 74 Dispensing with chemotherapy would be welcome but there are open questions: will there be unintended delivery to cells other than hematopoietic stem cells? How long does the “cure” last with this approach and how frequently would re-dosing be needed?
Conclusions
The advent of gene therapy for SCD marks a transformative shift in the landscape of treatment options for this debilitating genetic disorder. Despite the availability of disease-modifying therapies like hydroxyurea, the only true curative option until recently was a HSCT, which remains inaccessible for many patients due to the challenges of finding suitable donors and the risks associated with the procedure. Autologous gene therapy has risen to the forefront as a promising cure, leveraging the ability to correct or modify the patient’s own genetic material.
Gene therapy approaches focus on three main strategies: correcting the underlying mutation in the β-globin gene; inducing HbF production; and adding functional globin genes. Technologies like CRISPR, base editing, and prime editing offer a means to precisely modify genes involved in disease pathology. Each technique has its own advantages and challenges, but collectively, they represent groundbreaking progress in the search for a cure.
As summarized earlier, a number of preclinical studies have demonstrated the potential of these therapies in both animal models and ex vivo human cell cultures. FDA-approved treatments like Lyfgenia and Casgevy are a testament to the potential of gene therapy in treating SCD. These therapies have demonstrated remarkable results, including the resolution of VOC, normalization of hemoglobin levels, and significant reductions in disease-related complications. The success of these treatments in clinical trials highlights the promise of gene therapy not only as a curative option but also as a long-term solution that offers patients the possibility of a life free from the debilitating symptoms of SCD. However, the path to these historic FDA approvals was not without challenges, including concerns about the long-term safety of gene therapies, such as potential off-target effects and the risks associated with the conditioning regimens used prior to therapy.
Despite the promise shown by these therapies in the clinic, several hurdles remain. One of the primary challenges is accessibility, as these treatments are complex, costly, and require specialized infrastructure. Additionally, the process of stem cell collection, genetic modification, and reinfusion can take many months, during which patients may continue to suffer from the symptoms of the disease. Furthermore, the need for long-term follow-up to assess the durability of treatment outcomes and manage any potential late-onset complications is critical. As the field evolves, efforts to improve manufacturing efficiency, reduce the cost of treatment, and expand access to care will be crucial in ensuring that these therapies reach a broader patient population.
Looking ahead, the future of gene therapy for SCD appears promising. Continued research is necessary to refine gene editing techniques, improve manufacturing processes, and enhance the safety and accessibility of these treatments. Moreover, as more clinical trials expand the patient populations eligible for gene therapy, including younger patients and those with complications like stroke, the scope of gene therapy’s impact will only grow. With ongoing advancements and a focus on improving patient outcomes, gene therapy could become the standard of care for SCD, offering hope to millions affected by this devastating disease.
Footnotes
Acknowledgment
Authors’ Contributions
All authors wrote sections of the article, and they all edited and approved of the final version. D.J. and H.B. created the figures; M.M. created the tables. All authors edited and approved the final version.
Author Disclosure Statement
D.J. served on an advisory board meeting for Vertex Pharmaceuticals in 2024.
Funding Information
No funding was received for this article.