
Abstract
Objective:
This study assessed the feasibility of safely achieving target glycated hemoglobin (A1C) of ≤7% by intensifying structured insulin titration regimens using inhaled human insulin (Exubera® [EXU] [Pfizer Inc., New York, NY] [insulin human (recombinant DNA origin)] inhalation powder) in patients with type 2 diabetes inadequately controlled on combination oral antidiabetes agents (OADs).
Methods:
In a randomized, open-label, parallel, 24-week multicenter trial, 107 type 2 diabetes patients with mean baseline A1C of 8.6% taking two or more OADs were randomized to adjust EXU before meals following either weekly office visits or more intense twice-weekly telephone/office consultations, using a simple but structured insulin titration algorithm seeking to attain specific premeal glucose levels. Primary outcome was the percentage of patients reaching A1C ≤7%; secondary measures were changes in A1C, eight-point self-monitored blood glucose values, postprandial glucose levels during a meal tolerance, and frequency of hypoglycemia.
Results:
A1C improved whether EXU was systematically titrated once (6.8%) or twice weekly (6.8%), and two-thirds of patients in both groups attained A1C ≤7% (69% and 67%, respectively). Relative to baseline, glucose profiles were reduced at all time points measured, and postprandial glucose levels during meal tolerance improved to a similar extent in both groups. There were 538 hypoglycemic events with twice-weekly and 343 with once-weekly EXU titration; other adverse events were similar between groups.
Conclusions:
Added to oral therapy, premeal inhaled insulin can safely achieve ≤7% A1C in most patients with type 2 diabetes inadequately controlled while taking two or more OADs if a once- or twice-weekly structured insulin titration regimen is used.
Introduction
Inhaled human insulin (Exubera® [EXU] [Pfizer Inc., New York, NY] [insulin human (recombinant DNA origin)] inhalation powder) was a short-acting prandial insulin previously approved by the Food and Drug Administration and European Medicines Agency for the treatment of adults with type 1 diabetes mellitus or T2DM 8 that was subsequently withdrawn. Earlier clinical trials with EXU in patients with T2DM suboptimally controlled on OADs demonstrated relatively large reductions in A1C levels, 9 –13 but they were not specifically designed to follow a structured insulin titration regimen to optimize control and did not assess different dosing algorithms. The current study examined a structured titration of EXU, either once or twice weekly, that sought targeted premeal glucose levels to achieve A1C ≤7% in patients with T2DM who are poorly controlled on two or more OADs.
Research Design and Methods
Enrolled patients were men or women, 18–80 years old, with T2DM for 6 months or more and treated for at least 3 months with two or more OADs (metformin, sulfonylureas, thiazolidinediones) at stable doses of at least half the maximum daily dose or the highest tolerated dose. Inclusion criteria included A1C levels between 7.5% and 10.0% and a body mass index between 25 and 40 kg/m2. Exclusion criteria included insulin therapy within 3 months prior to screening, two or more severe hypoglycemic events within 6 months prior to screening, a history of smoking within 6 months prior to screening, significant pulmonary disease (moderate to severe asthma or chronic obstructive pulmonary disease), abnormal pulmonary function test (PFT), current use of α-glucosidase inhibitors or meglitinides, or the use of other agents affecting glycemic control (including systemic glucocorticoids and nonselective β-sympathetic blockers).
Study design
This randomized, open-label, parallel group (two-arm) study was performed at 18 centers in the United States between November 29, 2005 and March 6, 2007. Eligible patients were randomized (1:1) to one of the two study treatment groups. The randomization was stratified by A1C obtained at screening (≤9.0%, >9.0%). Randomization, performed centrally by a computer-generated scheme, used real-time block rand code distribution to balance treatments by center. The study was conducted in accordance with the Declaration of Helsinki and approved by local ethical review committees, and all patients provided informed written consent.
Study protocol and treatment
Patients were randomized to one of two arms. In one arm (Group 1), dose titration of EXU was performed following once-weekly contact (telephone or office visit) with the patient, whereas in the other arm (Group 2) dose titration was performed following twice-weekly contact. During the initial 4 weeks after randomization, patients in Group 1 had a telephone visit approximately once a week for study weeks when there was no scheduled clinic visit. Telephone visits were scheduled 7 ± 2 days following the most recent clinic visit. Patients in Group 2 had telephone visits approximately once a week for weeks when there was a scheduled clinic visit and approximately twice a week for weeks when there was no scheduled clinic visit. Telephone visits were scheduled 4 ± 1 days following the most recent clinic or telephone visit. During subsequent weeks, subjects in both groups who had reached a maintenance dose of EXU had a scheduled telephone visit during Weeks 6, 10, 14, 18, and 22. These telephone visits were scheduled approximately halfway between scheduled site visits. Subjects who had not reached the maintenance dose of EXU by Week 4 continued the initial 4-week telephone visit schedule until they reached the maintenance dose. Maintenance dose was defined as EXU dose titration not required for any of the three meals for two consecutive dosing assessments.
All patients were instructed to perform self-monitored blood glucose (SMBG) testing at least four times daily (prebreakfast [fasting], prelunch, predinner, and bedtime) and whenever patients experienced symptoms that might be related to a hypoglycemic event and to record the results. Hypoglycemia was defined as typical symptoms without glucose measurement but prompt resolution with food intake, typical symptoms with glucose concentrations of ≤59 mg/dL (≤3.3 mmol/L), or any glucose measurement of ≤49 mg/dL (≤2.7 mmol/L). For classification as severe hypoglycemia, all of the following criteria had to be met: (1) the subject was unable to treat himself or herself, (2) the subject exhibited neurologic symptoms (memory loss, confusion, uncontrollable or irrational behavior, difficulty in awakening, seizure, or coma), and (3) blood glucose ≤49 mg/dL (≤2.7 mmol/L) or, if not measured, the clinical manifestations were reversed by oral carbohydrates, subcutaneous glucagon, or intravenous glucose.
In both arms, OAD therapy was continued at the same dosages as at screening. The maximum daily doses of OADs allowed were 8 mg for glimepiride, 20 mg for glipizide or glyburide, 6 mg for micronized glyburide, 2,550 mg or 2,000 mg extended release formulation for metformin, 8 mg for rosiglitazone, 30 mg for pioglitazone, or fixed combinations of 20 + 2,000 mg for glyburide (or glipizide) plus metformin or 8 + 2,000 mg for rosiglitazone plus metformin. EXU was inhaled 10 min before each meal. The starting EXU dose was 0.05 mg/kg of body weight per meal (based on a total dose of 0.15 mg/kg divided equally among breakfast, lunch, and dinner). No carbohydrate counting was used in this study. Subsequent dose titrations were made by the study site based on the patient's three preceding SMBG readings and the EXU dose titration algorithm. Prebreakfast, prelunch, and predinner goals were 80 to <110 mg/dL (4.4 to <6.1 mmol/L), and EXU was increased by 1 mg (∼3 IU) if the blood glucose trend was >110 and <250 mg/dL (>4.4 and <13.9 mmol/L) and by 3 mg if the blood glucose trend was >250 mg/dL (>13.9 mmol/L). The dose was reduced by 1 mg if the blood glucose trend was <80 mg/dL. A trend was made up of two-thirds or more of the preceding SMBG values for a particular meal. Prebedtime goals were higher to prevent nocturnal hypoglycemia. If patients had a preprandial SMBG value ≤69 mg/dL (≤3.8 mmol/L), they were to decrease their EXU dose by 1 mg for that meal only and eat immediately. Likewise, if a preprandial SMBG was >300 mg/dL (>16.7 mmol/L), they were to increase the EXU dose for that meal by 1 mg. Prebreakfast adjustment were based on prelunch SMBG values, and the values at dinner determined the prelunch adjustments. Predinner adjustments were based on the prebedtime and fasting blood glucose values.
Patients visited the research site at screening (Week −2), at baseline (Week −1), at randomization (Week 0), and at Weeks 1, 2, 4, 8, 12, 16, 20, and 24. They met with a nutritionist at Week −1 and Week 8 for counseling on an appropriate diet that included three meals per day (breakfast, lunch, and dinner) with or without additional snacks. Meals were to be at least 3 h apart. Blood was collected and analyzed at a central laboratory for A1C and fasting plasma glucose (FPG). A1C was measured at Weeks −2, 4, 8, 12, 16, 20, and 24; FPG was measured at Weeks −1, 4, 8, and 24. SMBG eight-point profiles (before and 2 h after breakfast, lunch, and dinner and at bedtime and 2–4 a.m.) were performed before Week 0 and prior to each subsequent office visit. Three-hour meal tolerance tests were conducted at Weeks −1, 8, and 24, where a 480-kcal liquid meal (Boost® high protein drink, Nestlé HealthCare Nutrition Inc., Minneapolis, MN) containing 66 g of carbohydrate, 32 g of sugar, 12 g of fat, and 30 g of protein was consumed within 15 min, and plasma glucose was measured at time 0 (fasting) and at 30, 60, 90, 120, and 180 min postdose. Weight and body mass index were measured at each visit. PFTs (forced expiratory volume in 1 s, diffusing capacity of the lung for carbon monoxide, and forced vital capacity) were performed by local laboratories at screening and at Weeks 12 and 24. Adverse events were recorded throughout the study.
Outcome measures
The primary outcome measure was the percentage of patients achieving A1C ≤7% at end point. Secondary outcome measures included change from baseline for A1C, FPG, postprandial plasma glucose, weight, and EXU dose; percentage of patients achieving A1C ≤6.5% or ≤6.0% at Week 24; time to maintenance EXU dose; and prevalence and severity of hypoglycemia.
Statistical analyses
For this pilot study, a sample size of 50 evaluable patients per arm would yield a 95% confidence interval (CI) with margin of error of +14% for the primary end point, the proportion of patients that attained A1C ≤7%. Overall, 120 patients would need to be randomized 1:1 to the two treatment groups in order to account for a 20% attrition rate.
The safety population included all randomized patients who received at least one dose of study medication, while the full analysis set (FAS) was a modified intent-to-treat population comprising all randomized patients who received one or more dose of study medication and had one or more post-baseline efficacy measurement. The last post-baseline measurement before discontinuation or completion of the study was considered the end point measurement (last observation carried forward [LOCF]). Primary outcome analysis was performed on the FAS and consisted of assessment by treatment group, via a two-sided 95% CI for the proportion of subjects with A1C ≤7.0%. Secondary efficacy analyses were performed on the FAS population. Prevalence/severity of hypoglycemia was summarized by crude event rate. A1C to-target end points were analyzed by logistic regression analysis (model consisting of treatment, pooled center, and baseline A1C).
Results
A total of 251 patients were screened, of which 108 were randomized (Fig. 1). One patient in Group 1 was not treated. The 107 patients treated comprised the safety group (53 in Group 1; 54 in Group 2). For efficacy analyses, the FAS consisted of 103 patients (51 in Group 1; 52 in Group 2). Of these 103 patients, 101 had an evaluable primary end point (49 in Group 1; 52 in Group 2). Sixteen patients in Group 1 and 13 in Group 2 withdrew from the study. Reasons for discontinuation included drug-related adverse events (7.5% in Group 1; 7.4% in Group 2), lost to follow-up (7.5% and 9.3%, respectively), and other (15.1% and 7.4%, respectively). Other included discontinuations for noncompliance and violation of protocol or the patient not meeting entrance criteria.
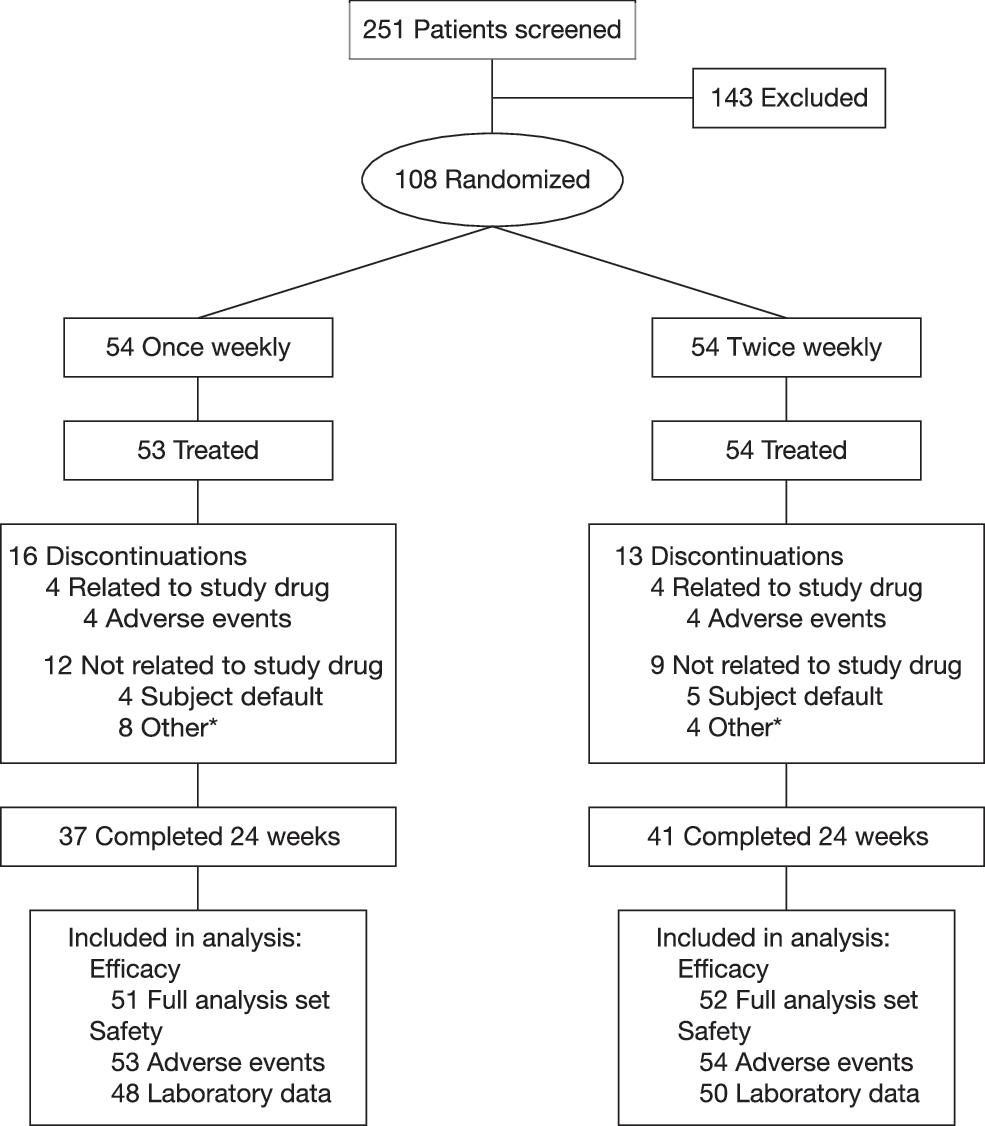
Patient disposition. *Includes discontinuations for noncompliance and violation of protocol or patient not meeting entrance criteria.
The baseline characteristics of each treatment group are shown in Table 1. Slightly more patients were male than female in both groups. Patients in Group 2 were slightly older and had diabetes slightly longer. Approximately 48% of all patients were on triple therapy, and about 43% were taking metformin plus sulfonylurea. The initial A1C averaged 8.6%.
Data are mean ± SD values. SU, sulfonylurea; TZD, thiazolidinediones.
Includes one patient on metformin plus nateglinide.
A1C
A1C levels declined at the same rate in both Group 1 and Group 2, stabilizing after 8–12 weeks (Fig. 2A). The mean A1C at Week 24 (LOCF) was 6.8% in both groups. The mean change from baseline in A1C was −1.76% and −1.72% in Groups 1 and 2, respectively (change from baseline between-treatment expected difference was −0.02% [95% CI, −0.29, 0.25], P = not significant) (Table 2). The A1C target of ≤7% was reached by 69.4% of patients in Group 1 and 67.3% in Group 2, with no differences in time course between the two groups. No differences were noted between the groups in the percentage of patients who achieved A1C ≤6.5% or ≤6.0% (Table 2).
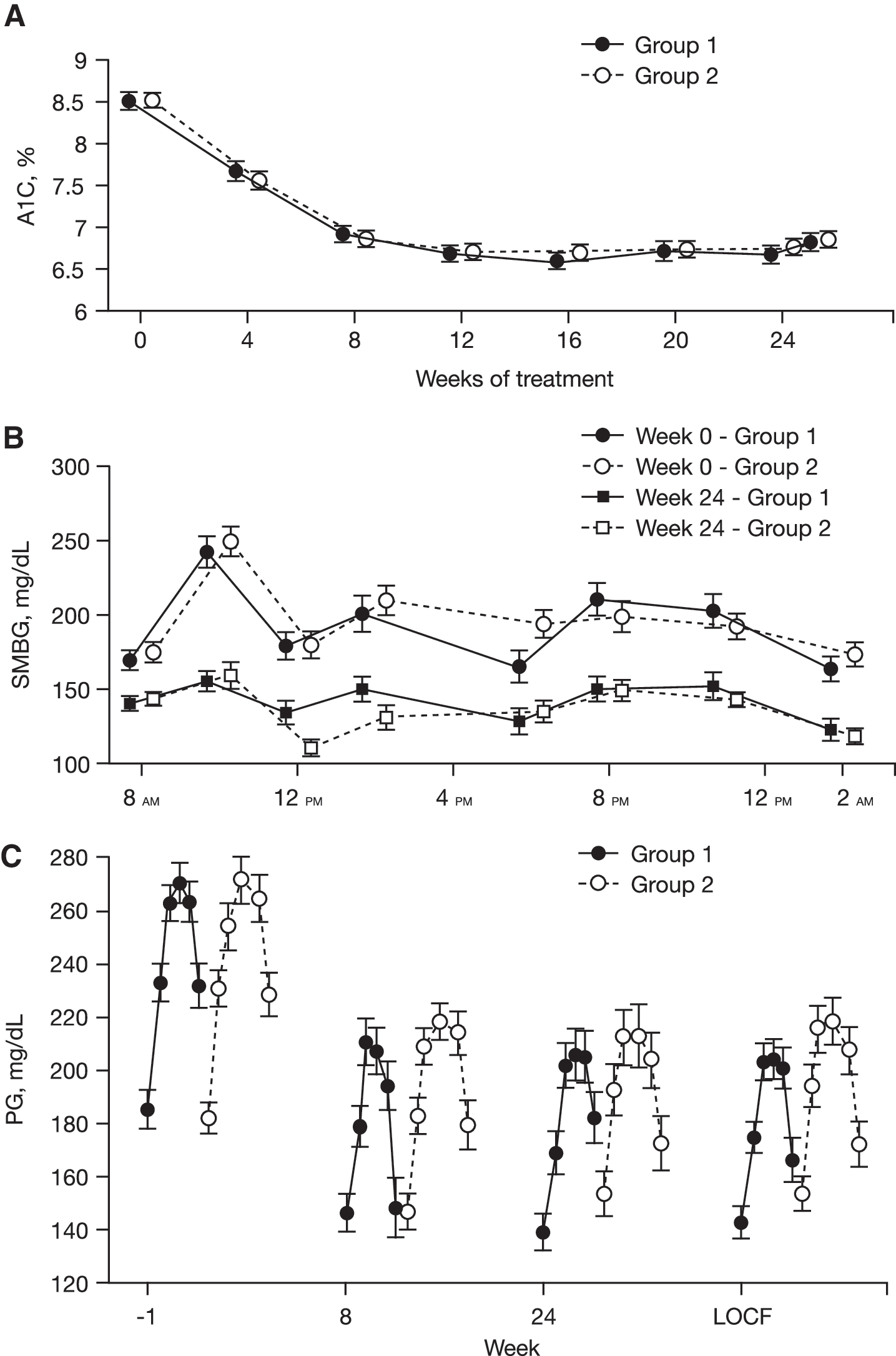
Effect of EXU on (
CFB, change from baseline.
Glycemic response, EXU dosage, and body weight
SMBG eight-point profiles at baseline and end point are shown in Figure 2B. Blood glucose values were statistically significantly reduced from baseline at all time points measured in both groups. There were no significant differences between groups except at prelunch, where the SMBG change from baseline in Group 2 was statistically significantly greater than in Group 1 (−70 ± 71 mg/dL vs. −44 ± 72 mg/dL; P = 0.012).
FPG and glucose excursions during the meal tolerance test were reduced in both groups at Weeks 8 and 24 (Fig. 2C). Within each group, the change from baseline glucose levels was statistically significant at all weeks for all time points. Between groups, significant differences in the change from baseline in plasma glucose were observed at Week 8 (P = 0.031) and Week 24 (P = 0.006), favoring once-weekly adjustments.
The daily dose of EXU was larger in Group 2 than in Group 1, but the difference was significant only through Week 8. The mean ± SD EXU dose at baseline was 11.6 ± 3.8 mg and 11.6 ± 4.8 mg in Group 1 and Group 2, respectively, and 25.3 ± 15.6 mg and 30.3 ± 20.2 mg at Week 24. The majority of patients in both groups required 12 or more weeks to reach a maintenance EXU dose. The mean ± SD increase in weight at Week 24 was similar in the two groups: 3.7 ± 3.6 kg in Group 1 and 3.5 ± 3.4 kg in Group 2 (between-treatment difference, 0.04 kg [95% CI, −1.36, 1.45], P = not significant).
Safety: hypoglycemia and adverse events
There were 538 and 343 hypoglycemic events or 2.1 and 1.4 events per subject-month in Group 2 and Group 1, respectively (Table 3). The number of patients with one or more event was comparable between groups, whereas only one patient in each group experienced severe hypoglycemia. The most common respiratory adverse event was cough (13.2% in Group 1; 9.3% in Group 2). PFT results were numerically similar between the two groups (Table 3).
DLCO, diffusing capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; IQR, interquartile range.
Conclusions
This study demonstrated that premeal EXU can be effectively intensified weekly following a simple but structured insulin titration algorithm that achieved the targeted goal of A1C ≤7% in approximately two-thirds of patients with T2DM uncontrolled on two or more OADs. Dose titrations were performed following either once- or twice-weekly telephone/office visits. The lack of difference whether titrations were done weekly or twice weekly indicated that good glycemic control could be obtained with simple, weekly adjustments to the EXU dose.
A greater percentage of patients in this study achieved target A1C than when EXU was added to either glibenclamide 12 or metformin. 13 For patients in these studies with baseline A1C ≤9.5%, mean change from baseline in A1C was similar (−1.84%) to that in the present study, but only 30–40% achieved A1C <7% compared with almost 70% who achieved A1C ≤7% in the current study. This difference probably reflects the difference in preprandial blood glucose targets, with a much tighter goal in the current study (80–110 mg/dL [4.4–6.1 mmol/L]) than in the earlier studies (80–140 mg/dL [4.4–7.8 mmol/L]). Adjustments to the EXU dose were made weekly in the earlier studies.
The percentage of patients achieving target A1C in this study is somewhat greater than or similar to that reported in other treat-to-target trials with injectable insulins in patients with T2DM who were uncontrolled on one or more OADs. A1C ≤7.0% was attained by 58.0% and 57.3% of patients receiving either insulin glargine or NPH insulin at bedtime, respectively, in the Treat-To-Target study. 14 Two-thirds of patients have been shown to attain A1C ≤7.0% with twice-daily biphasic insulin aspart 70/30 but at the expense of higher insulin doses, more hypoglycemia, and greater weight gain than basal insulin glargine 15 or with the more complex regimen of insulin glargine at bedtime plus mealtime lispro. 16
In the present study, safety-related findings were comparable between groups and supported previous evidence that EXU is well tolerated in patients with T2DM. 10 –13,17,18 Added to oral therapy, premeal inhaled insulin can safely achieve A1C ≤7% in a majority of overweight patients with T2DM with A1C between 7.5% and 10.0% inadequately controlled on two or more OADs if a once- or twice-weekly structured insulin titration regimen is used.
Addendum
On October 18, 2007 Pfizer Inc. announced that it would cease marketing Exubera because it did not meet customers' needs or financial expectations.
On April 9, 2008 Pfizer Inc. announced that it was updating the Exubera product insert to include the following statement: “In studies of Exubera in people with diabetes, lung cancer occurred in a few more people who were taking Exubera than in people who were taking other diabetes medicines. All of the people in these studies who developed lung cancer used to smoke cigarettes. There were too few cases to know if the lung cancer was related to Exubera.”
Footnotes
Acknowledgments
This study was sponsored by Pfizer Inc. Medical writing support was provided by Tom Claus, Ph.D., of PAREXEL throughout the development of the manuscript. However, the intellectual content of the manuscript was directed by the authors; all authors were involved in the study design and analysis, provided critical intellectual input into the manuscript, and signed off on the final content. Editorial support was funded by Pfizer Inc. We would like to acknowledge the principal investigators involved in this study: Enrico Cagliero, M.D., Massachusetts General Hospital, Boston, MA; William T. Cefalu, M.D., Pennington Biomedical Research Center, Baton Rouge, LA; Richard B. Christensen, M.D., Humphreys Diabetes Education Center, Boise, ID; James Desemone, M.D., Albany Medical Center, Albany, NY; Thomas M. Flood, M.D., Georgia Center for Diabetes, Atlanta, GA; David M. Gorson, M.D., Bennington, VT; Priscilla A. Hollander, M.D., Baylor University Medical Center, Dallas, TX; Sam Lerman, M.D., The Center for Diabetes and Endocrine Care, Hollywood, FL; Ruchi Mathur, M.D., USC Health Care Consultation Center II, Los Angeles, CA; Sam S. Miller, M.D., Sam Clinical Research Center, San Antonio, TX; Fernando Ovalle, M.D., University of Alabama at Birmingham, Birmingham, AL; William A. Petit, Jr., M.D., New Britain General Hospital, New Britain, CT; Terrance A. Riske, M.D., Hayden Lake Family Physicians, Hayden, ID; Julio Rosenstock, M.D., Dallas Diabetes and Endocrine Research Center, Dallas, TX; Sergio F. Rovner, M.D., E.P. Premier Medical Group, El Paso, TX; Mindy J. Sotsky, M.D., Soundview Research Associates, Norwalk, CT; Guillermo E. Umpierrez, M.D., Emory University, Atlanta, GA; and Ruth S. Weinstock, M.D., SUNY Upstate Medical University, Syracuse, NY.
Author Disclosure Statement
P.A.H., W.T.C., and J.R. have received grant support for clinical research and honorarium for having served on Pfizer Scientific Advisory Boards. M.M and D.L. are employees of Pfizer Inc.
This study is registered at http://clinicaltrials.gov as trial number NCT00246623. Its protocol number is A2171067.