
Abstract
Diabetic cardiomyopathy is a distinct entity in humans. It leads to ventricular dysfunction independent of and additive to coronary artery disease and hypertension. Clinical and experimental studies have pointed to the role of metabolic derangements in the development of diabetic cardiomyopathy. Altered insulin signaling in diabetes leads to decreased myocyte glucose uptake and utilization, associated with an increased concentration of free fatty acids. This results in decreased glucose oxidation and increased fatty acid oxidation. Fatty acids increase mitochondrial oxygen consumption for ATP production and stimulate the uncoupling proteins in mitochondria. These proteins decrease the mitochondrial protein gradient, leading to fall in ATP production. The resultant defect in myocardial energy production impairs myocyte contraction and diastolic function. This is the hallmark of diabetic cardiomyopathy at earlier stages. In later stages diabetes impairs the myocyte ischemic defense mechanism, leading to increased cardiovascular morbidity and mortality. Other factors contributing toward causation of diabetic cardiomyopathy are collagen accumulation leading to reduced myocardial compliance, accumulation of advanced glycation end product–modified extracellular matrix proteins with subsequent inelasticity of vessel walls and myocytes, abnormal myocardial calcium handling leading to altered mechanics, endothelial dysfunction, cardiac autonomic neuropathy, and impairment of ischemic preconditioning. Trimetazidine acts a metabolic switch, favoring glucose over free fatty acids as the substrate for metabolism in cardiac myocytes.
Introduction
Normally the heart uses nonesterified or free fatty acids as its primary energy substrate during aerobic perfusion at normal workloads and increasingly relies on glycolysis and pyruvate oxidation during periods of ischemia and increased stress. Because of reduced glucose transport into cardiac myocyte, the heart in diabetes can have an exaggerated impairment in ATP generation by glycolysis during ischemia. Accumulated free fatty acids and their oxidation products are toxic to myocardial cells, contributing to diabetic cardiomyopathy.
We summarize here the current concept of cardiac abnormalities due to metabolic perturbations that accompany diabetes. The aim is to optimize present and future therapeutic strategies to influence myocardial function favorably.
Effect of Diabetes on Cardiac Energy Metabolism
Fatty acid oxidation in the mitochondria serves as the main myocardial energy substrate in a resting heart. There is also significant utilization of glucose for cellular energy (ATP) production during periods of increased demand. Insulin resistance in diabetes results in decreased utilization of carbohydrates regardless of coronary artery disease and despite normal basal blood flow. 9 Stimulation of lipases leads to increased levels of free fatty acids, upon which the myocardium of the heart with diabetes relies increasingly for energy production by oxidation (Fig. 1).
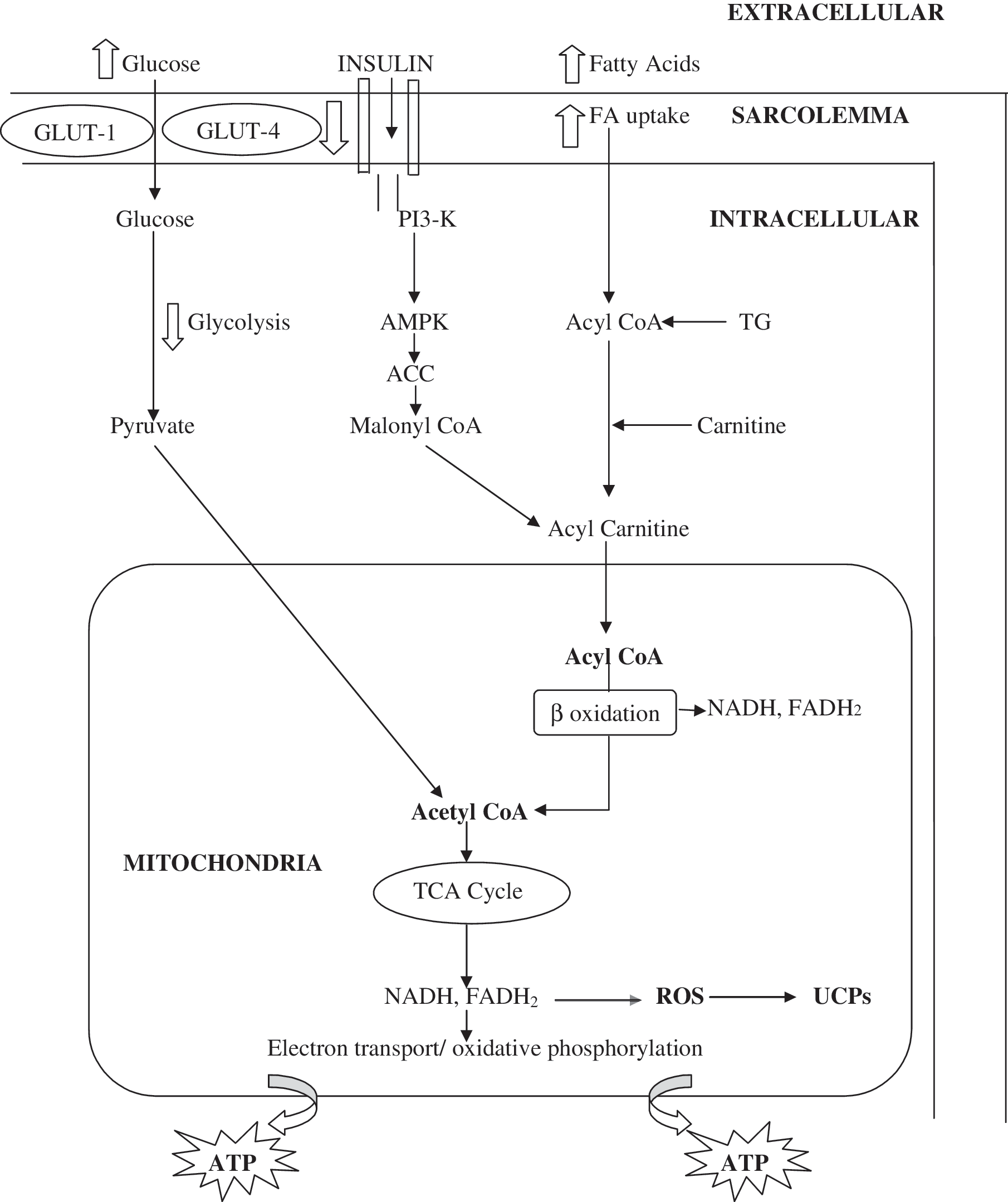
Schematic diagram showing cardiac myocyte energy metabolism. In the heart of a patient with diabetes, high concentrations of fatty acids and insulin resistance lead to decreased glucose oxidation and increased fatty acid oxidation. ACC, acetyl-coenzyme A carboxylase; Akt, protein kinase B; AMPK, 5-AMP-activated protein kinase; CoA, coenzyme A; FA, fatty acid; GLUT, glucose transporter; PI3K, phosphatidylinositol 3-kinase; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; TG, triglycerides; UCP, uncoupling protein.
Fatty acids are less efficient fuel, and their oxidation in mitochondria for ATP production results in higher mitochondrial oxygen consumption. 10 Fatty acids up-regulate the expression and activity of uncoupling proteins (UCPs) in the inner membrane of mitochondria. The UCPs act as transport proteins and belong to the anion mitochondrial carriers. One such example is UCP 1 in brown adipose tissue. 11 It converts energy stored within the mitochondrial proton electrochemical potential gradient to heat, leading to decreased conversion of ADP to ATP. In contrast, UCP 2 is expressed widely, and UCP 3 is expressed preferentially in skeletal muscle. They are also located in the heart. 12 UCP 1 plays an important role in regulating heat production during cold exposure, but the biological functions of UCP 2 and UCP 3 are not known. Possible functions include (1) control of adaptive thermogenesis, (2) control of reactive oxygen species production by mitochondria, (3) regulation of ATP synthesis, and (4) regulation of fatty acid oxidation. 13 UCP levels correlate routinely with fasting concentrations of plasma free fatty acids and inversely with the concentration of insulin-responsive glucose transporter-4 protein. 12 Rolfe and Brand 14 estimated that proton leak accounts for 26% of resting energy expenditure.
Fatty acids trigger insulin resistance by inhibiting insulin signaling pathways. 15 Binding of insulin to insulin receptors stimulates tyrosine kinase activity, leading to autophosphorylation of the insulin receptor and insulin receptor substrate (IRS) molecules. Fatty acyl-coenzyme (CoA) induces activation of atypical protein kinase C, a serine-threonine kinase that phosphorylates and subsequently activates inhibitor of κB kinase, 16 which in turn phosphorylates serine residues on IRS, inhibiting its ability to bind SH2 domains of the p85 regulatory subunit of the lipid kinase, phosphatidylinositol-3-kinase. This results in impaired insulin signal transduction. Finally, the recruitment of glucose transporter-4 transporters to plasma membrane is down-regulated, there by compromising glucose uptake. This mechanism is active in skeletal muscle and adipose tissue. However, it is likely that similar mechanisms play a role in myocardium.
In addition to insulin, 5-AMP-activated protein kinase (AMPK) is also an important regulator of glucose metabolism in the heart. 17 AMPK acts as a cellular fuel gauge, by shutting off energy-requiring processes and stimulating energy-yielding processes during times of metabolic stress. AMPK can mimic the effects of insulin on myocardial glucose uptake and glycolysis. 18 However, unlike insulin, AMPK activation is associated with an acceleration of fatty acid oxidation in the heart, which in turn can lead to a decrease in glucose oxidation. 19 AMPK also inhibits fatty acid and protein synthesis, which is apparently opposite to the action of insulin. AMPK inactivates acetyl-CoA carboxylase in heart and decreases the concentration of malonyl-CoA. The latter normally inhibits the entry and subsequent oxidation of long-chain fatty acids in the mitochondria. 20 Insulin antagonizes AMPK activation only in the absence of free fatty acids. 21 Therefore, interaction between insulin and AMPK increases the malonyl-CoA concentration and consequently limits fatty acid oxidation while facilitating glucose oxidation. Insulin-induced regulation of AMPK is blunted by high plasma concentrations of free fatty acids in diabetes. 22
Furthermore, AMPK has a differential role in the activation of components in the cardiac insulin signaling pathway. On one hand, AMPK activation improves insulin sensitivity by (1) activating insulin-induced phosphorylation of glycogen synthase kinase 3β, p70 S6 kinase, and IRS1 and (2) suppressing mammalian target of rapamycin/S6 kinase–mediated negative feedback regulation of insulin signaling. At the same time AMPK promotes insulin resistance by (1) inhibiting the insulin-induced stimulation of IRS1-associated phosphatidylinositol 3-kinase activity and (2) activates protein kinase C, leading to down-regulation of the insulin signaling pathway. 23
Ventricular Dysfunction in Diabetes
Diabetes leads to accumulation of lipid intermediates in cardiac myocytes. 24 Long-chain acyl-CoA esters synthesized at the outer mitochondrial membrane from C16 and C18 fatty acids facilitate opening of cell membrane K+-ATP channels by reducing ATP sensitivity. 25 The resulting shortening of action potential would lead to a reduction of Ca2+ influx across the sarcolemma 25 with subsequent reduction in myocardial contractility. Insulin-dependent diabetes also impairs sarcoplasmic reticulum calcium pump activities, which reduces the rate of calcium removal from cytoplasm of cardiac myocytes, contributing to increased diastolic stiffness. 26 In type 2 diabetes, there is a disproportionate increase in left ventricular mass. 6,27
The cardiac sarcolemmal Na+–H+ exchanger (NH1 isoform 1) is involved in control of myocellular pH control and in linking of cardiac metabolism to ionic homeostasis. NH1 favors left ventricular hypertrophy in type 2 diabetes during impaired myocardial perfusion. 28
Accumulation of advanced glycation end products modifies the extracellular matrix components, causing vessel wall inelasticity and impairment of myocardial function as well. Serum levels of advanced glycation end products correlate with prolongation of isovolumetric relaxation time as assessed by Doppler echocardiography. 29 The opening of myocardial ATP-sensitive potassium channels is an essential factor in ischemic preconditioning in human myocardium. Both diabetes itself and the usage of sulfonylureas (which block these potassium channels) appear to eliminate this beneficial effect of ischemic preconditioning. 30
Excessive reliance on fatty acid oxidation has two effects. It results in higher mitochondrial oxygen consumption and stimulation of UCP 2 and UCP 3, leading to a lowered mitochondrial proton gradient without ATP generation with a subsequent decrease in myocardial ATP production. Increased delivery of reducing equivalents like FADH2 and NADH from fatty acid oxidation leads to increased generation of mitochondrial reactive oxygen species, which in turn activates UCP. 31 Altered myocardial energetic precede the echocardiographic alteration of cardiac function. 32 These alterations are negatively correlated with fasting plasma free fatty acid concentrations.
Therapeutic Implications
The basic aim of the treatment is to reset the metabolic switch from free fatty acids toward more utilization of glucose for efficient energy production. 33 Trimetazidine acts as a metabolic modulator, inhibiting the long-chain 3-ketoacyl-CoA thiolase, which is the last enzyme involved in mitochondrial fatty acid β-oxidation. 34 Although the myocardial fatty acid oxidation was decreased by 10%, 35 it could significantly increase the glycolytic flow through pyruvate dehydrogenase by the Randle mechanism 36 and by anaplerosis of pyruvate to oxaloacetate and malate. 37
Insulin resistance in type 2 diabetes leads to increased myocardial fatty acid uptake 38 and decreased myocardial glucose uptake, 39 worsening the energy depletion state. Also, trimetazidine improves insulin resistance. 40 β-Blockers are also known to improve insulin resistance 41 and induce the shift from fatty acids to glucose oxidation. 42 So trimetazidine and β-blockers act synergistically.
Other possible modes of action of trimetazidine include the following: (1) Cardiomyocyte apoptosis could be inhibited. 42 (2) Because fatty acid metabolism-induced activation of AMPK inhibits protein synthesis and myocyte growth, by inhibiting fatty acid metabolism, trimetazidine might limit AMPK activity and permit greater myocyte growth. 43 (3) The metabolic profile is improved with an increase in high-density lipoprotein by 11%. 35 (4) By raising phosphocreatinine levels, trimetazidine increases the availability of phosphate and subsequent ATP. Overall improvement in ejection fraction occurs in heart failure with diabetes. 44
Ranolazine is a selective inhibitor of the late sodium current relative to the peak sodium channel current, and via this mechanism, it may decrease sodium-dependent intracellular calcium overload during ischemia and reperfusion. 45 It does play a role when ischemia accompanies diabetes. Newer measures improving coupling between fatty acid delivery and oxidation to limit production of reactive oxygen species could be of help. One such measure is inhibiting poly(ADP-ribosyl) polymerase (PARP), which acts as a DNA-nick sensor enzyme. 46 In endothelial cells, hyperglycemia-induced overproduction of mitochondrial superoxide causes DNA strand breaks, leading to activation of PARP, which inhibits glyceraldehyde 3-phosphate dehydrogenase. This leads to the accumulation of glucose and other glycolytic intermediates prior to their entry into the Krebs cycle. These intermediaries activate several major transducers of hyperglycemic damage (polyol pathway, advanced glycation end products formation, and protein kinase Cβ activation). 47 In addition to the direct cytotoxic pathway regulated by DNA injury, PARP also modulates the course of cardiovascular inflammation and injury by regulating the activation of nuclear factor-κB 46 and inducing overexpression of endothelin-1 and endothelin receptors. 47 Blocking PARP activity with competitive PARP inhibitors provides a “magic bullet” approach as it blocks activation of all the major pathways thought to mediate tissue damage in diabetes cardiomyopathy. 48
Conclusions
Defective cardiac energy metabolism impairs cardiac contractile function leading to the hallmark diastolic dysfunction. 49 Glucose oxidation is decreased, and fatty acid oxidation is increased. Fatty acids increase mitochondrial oxygen consumption and decrease the mitochondrial protein gradient with a resulting decrease in ATP production. The resultant defect in myocardial energy production leads to diastolic dysfunction. This further limits the ability of myocardium to withstand ischemia. 50 Trimetazidine shifts the energy substrate preference to glucose and also has positive effects on insulin sensitivity and glucose homeostasis. Ranolazine can be of help when ischemia accompanies diabetes.
Footnotes
Author Disclosure Statement
No competing financial interests exist.