
Abstract
Introduction:
For people with type 2 diabetes (T2D), optimal glycemic control is critical. Digital health interventions are a practical approach to improve T2D self-management. This study explored the safety and functionality of a novel clinical decision-support (CDS) application for individualized dosing of insulin efsitora (efsitora), a once-weekly basal insulin receptor agonist.
Participants and Methods:
This was a 16-week, multicenter, open-label early feasibility study in adults with T2D with or without basal insulin. Investigators requested an efsitora dose from the CDS algorithm and either overrode or accepted the recommendation. Dose recommendation overrides (primary endpoint), finger-stick glucose, blinded continuous glucose monitoring metrics, and hypoglycemic events were evaluated.
Results:
Two sequential cohorts consisted of 68 participants; each cohort included insulin-naïve and basal-switch participants. In both cohorts, mean glycated hemoglobin (HbA1c) for basal-switch participants ranged from 7.9% to 8.5%. Mean HbA1c for insulin-naïve participants ranged from 8.1% to 8.3%. CDS dosing recommendation overrides occurred for 0.7% of injections for basal-switch participants and for 1.0% of injections for insulin-naïve participants in Cohort 1. For Cohort 2, overrides occurred for 1.3% of injections for insulin-naïve participants, with no overrides for basal-switch participants. HbA1c was significantly reduced (P < 0.05) from baseline to Week 16 in both subgroups for both cohorts. The proportion of participants with fasting blood glucose within the targets increased from baseline to Week 16 in both subgroups for both cohorts. No level 3 hypoglycemia was observed.
Conclusions:
The novel CDS algorithm showed promising clinical performance and favorable investigator confidence as determined by a low rate of dose overrides.
Introduction
With approximately 25% of people with diabetes treated with insulin in the United States, 1 optimal glycemic control is critical to reduce the risk of acute and long-term complications. 2 However, many people with diabetes experience suboptimal glycemic control, with only 30% of those treated with basal insulin therapy adhering to treatment. 3
Insulin efsitora alfa (efsitora) is an insulin receptor agonist designed for once-weekly administration and has been investigated in a number of Phase 3 clinical trials, all of which have met the primary endpoint. 4,5 Efsitora has a pharmacokinetic (PK) half-life of approximately 17 days. 4 A once-weekly basal insulin has the potential to increase willingness to initiate, and improve compliance with, an insulin therapy in people with type 2 diabetes (T2D). 6 –8 However, titrating the dose of an insulin with a half-life of approximately 17 days requires patients and prescribers to learn the novel dosing regimen of once-weekly insulin and the resultant effects of glycemia. 6
Self-management, education, and health care professional support are recommended to ensure that people with diabetes have the tools needed for successful management of their condition. 9 Digital health interventions are a feasible and effective approach to self-management of T2D. 10,11 The investigational efsitora clinical decision-support (CDS) dosing strategy is a novel investigational digital alternative to conventional dosing algorithms that incorporates fasting blood glucose (FBG) trends, prior insulin doses, population PK modeling, and hypoglycemic events. The CDS algorithm was developed based on the PK and pharmacodynamics (PD) of efsitora and was designed to bring FBG values into clinical range with efsitora titration while limiting the risk of hypoglycemia. The CDS is a proportional derivative algorithm with adaptive gain and an internal model of insulin activity to support insulin titration based on efsitora PK/PD that recommends changes in dose based on FBG levels relative to the target and how quickly FBG levels change over time.
The aim of the current study was to evaluate the feasibility and safety of the CDS dosing strategy via the number of medically appropriate clinician overrides of dosing algorithm recommendations and collection of adverse events (AEs) in addition to measures of glycemic control.
Participants and Methods
Study design
This was a multicenter, single-arm, open-label, early feasibility study conducted with two sequential cohorts (Cohort 1 and Cohort 2) and included participants with T2D either previously treated with basal insulin (basal switch) or who were insulin naïve. Based on the outcomes in Cohort 1, the CDS algorithm was modified for Cohort 2 (Supplementary Table S1). For both cohorts, the study design consisted of four periods: a screening period of up to 4 weeks; a lead-in period of approximately 2 weeks; a treatment period of approximately 16 weeks; and a safety follow-up period of approximately 5 weeks (Fig. 1). Continuous glucose monitoring (CGM) data were collected from all participants throughout the lead-in, treatment, and safety follow-up periods using the Dexcom G6 Pro (San Diego, CA). Both investigators and participants remained blinded to the CGM data during the lead-in and treatment periods but were only unblinded to investigators during the safety follow-up period. In addition, screening laboratories were conducted during the lead-in period, and nonstudy diabetes therapy was uptitrated during the safety follow-up period. Participants monitored their FBG using glucometers (CONTOUR® NEXT ONE blood glucose monitor, Parsippany, NJ). The study was performed in compliance with the consensus ethical principles derived from international ethics guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Council on Harmonisation Good Clinical Practice guidelines, applicable International Organization for Standardization (ISO)14155 Clinical Investigation of Medical Devices for Human Subjections—good clinical practice guidelines, and applicable laws and regulations. All participants provided written informed consent before participating in the study.

Study design for both Cohort 1 and Cohort 2.
Study population
Eligible participants were 18 to 75 years of age, had a body mass index of 20–45 kg/m2, weighed between 52 kg and 198 kg, and had no significant weight gain or loss (≥5%) in the past 3 months. All participants were required to be able to use a smartphone. For Cohort 1, all participants were required to have a baseline glycated hemoglobin (HbA1c) value of 7.0%–9.5%, as determined by a central laboratory at screening. Participants in the insulin-naïve subgroup of Cohort 1 were diagnosed with T2D and treated with a stable dose of metformin (alone or in combination with a stable dose of dipeptidyl peptidase-4 inhibitor [DPP-4i] and/or a sodium–glucose cotransporter-2 inhibitor [SGLT-2i]) for ≥3 months before screening. For Cohort 2, all participants were required to have a baseline HbA1c value of 7.0%–10.0%, as determined by a central laboratory at screening. Participants in the insulin-naïve subgroup of Cohort 2 were diagnosed with T2D and treated with a stable dose of ≥1 antidiabetic medication (metformin, a DPP-4i, sulfonylurea, and/or an SGLT-2i) for ≥3 months before screening. Participants were included in a basal-switch subgroup in both cohorts if they were diagnosed with T2D according to the World Health Organization criteria; were treated with basal insulin for ≥6 months; had a stable regimen of insulin glargine (U100 or U300), insulin detemir, or insulin degludec (U100 or U200) with or without oral antihyperglycemic medication therapy (DPP-4i, SGLT-2i, biguanides, alpha-glucosidase inhibitors, sulfonylureas) before screening for ≥3 months; and were willing to continue stable dosing throughout the study. Participants in the basal-switch subgroup were included if they were receiving ≥10 U of basal insulin per day and ≤1.5 U/kg of basal insulin per day.
Participants were excluded from the study if they had a history of ≥1 episode of ketoacidosis or hyperosmolar hyperglycemic state/coma requiring hospitalization within 6 months of screening or had any episodes of severe hypoglycemia and/or hypoglycemia unawareness within 6 months of screening.
Device description
The investigational device, CDS, is a novel investigational dosing-algorithm software system designed to enable clinical evaluation of the dosing algorithm through weekly dose recommendations of efsitora (Supplementary Fig. S1). The CDS comprised four integrated components as follows: Data Acquisition, which is supported by CONTOUR® NEXT ONE blood glucose monitor (BGM), as per the manufacturer’s instructions. First, blood glucose values were captured by the participant and then transmitted from the BGM to the CONTOUR DIABETES iPhone application (app). Once blood glucose values were available in the mobile app, participants were able to annotate relevant values with additional details, which were reviewed by the investigator and provided input to the algorithm. Before each dose recommendation, participants emailed the application blood glucose data file to a designated email address for use within the CDS. The Data Hub provided services to ingest, store, and manage data related to study participants, the Investigator Portal, and the CDS software library. The Investigator Portal (Indianapolis, IN) was a web-based user interface that provided the site investigator with the capability to review and apply clinical judgment as to whether BGM data should be excluded or included in the basal dose calculation. Upon receipt of the dose recommendation, the investigator was able to accept the recommended dose or override it for safety reasons before actual dose administration to the participant. Any dose override was captured in the CDS. The CDS software library comprised three modules: (1) one-time introductory (loading) dose, (2) safety, and (3) titration, which were used to calculate weekly recommended efsitora doses.
Algorithm description
The one-time introductory dose (Week 1) and the subsequent titration doses were calculated via the Investigator Portal for both insulin-naïve and basal-switch participants with T2D (Supplementary Fig. S2). In this study, the one-time introductory dose was determined via an extensive lookup table based on current body weight and HbA1c value for insulin-naïve participants, and HbA1c value and baseline basal insulin dose for basal-switch participants. Both the insulin-naïve and basal-switch participants shared the same safety and titration modules that when engaged recommended a dose reduction.
The safety module identified whether any hypoglycemic events occurred between visits in Cohort 1 and if any were recorded within 7 days before the current visit in Cohort 2. If there were no safety concerns associated with hypoglycemia, the CDS progressed to the titration module. To reach a predicted insulin concentration level, the recommended dose was calculated based on the active titration phase and the maintenance dose was calculated from the estimated insulin action phase. The active titration phase made dose adjustments using a proportional derivative controller, which was designed to bring FBG values into a predefined titration target range. To determine the dose adjustment, the controller balanced the difference between the current FBG and the control target as well as the direction of the glucose trend. From the second titration dose, the CDS was allowed to consider recommending a boost dose (a substantial dose increase intended to help the participant reach a new steady state more quickly but was not intended to be a long-term steady-state dose) if all safety prerequisites were fulfilled (absence of prior hypoglycemia, an adequate number of FBG measurements, and mean and median checks of FBG values). The maintenance dose based on the estimated insulin action phase was designed to continue with the most recently administered dose amount to maintain insulin activity if the participant’s FBG values were in the target range without any hypoglycemia.
Treatment protocol
In both cohorts, efsitora was administered once weekly as a subcutaneous injection from Week 1 through Week 16 for a total of 16 doses. Titration and administration were conducted at the investigation site by the investigator. At each visit, investigators reviewed the BGM data via the investigator dashboard, including daily FBG and any hypoglycemic event information. Dose recommendations for each efsitora dose were then calculated by the investigator using the CDS investigational device.
Outcome measures
The primary objective of this study was to evaluate the safety of the CDS by assessing the number of medically appropriate clinician overrides of the dosing algorithm recommendation and the number of AEs. All overrides were determined to be medically appropriate. The secondary objective was to evaluate other measures of glycemic control and time in range by assessing change in FBG and HbA1c from baseline to Week 16; proportion of participants reaching HbA1c <7.0% at Week 16; frequency, severity, and timing of hypoglycemic events; number of overrides of dose recommendation; proportion of participants in Cohort 1 reaching a target FBG of 80–100 mg/dL (4.4–5.6 mmol/L) at Weeks 8 and 16; and proportion of participants in Cohort 2 reaching a target FBG of 80–100 mg/dL and 80–120 mg/dL (4.4–6.7 mmol/L) at Weeks 8 and 16. The exploratory objective was to evaluate the functionality and design of the CDS by assessing CGM time in ranges (70–180 mg/dL, <54 mg/dL, <70 mg/dL, and >180 mg/dL over the final 2 weeks of treatment), time to achieve a stable administered dose (defined as the first 3 weeks of static dosing), mean and mean change in insulin dose over time, and time to achieve a final constant dose (defined as the final dose with no further changes).
Statistical analysis
All functionality and safety analyses were conducted on the modified intent-to-treat population, which included all participants who received at least one dose of study treatment.
All tests were conducted at a two-sided alpha level of 0.05. The number of medically appropriate clinician overrides of the dosing algorithm recommendation was summarized by visit and during the overall treatment period. Analyses of AEs were descriptive and included all data collected during the treatment period. Change from baseline in HbA1c was analyzed using the Wilcoxon signed-rank test. Analyses of other continuous variables, including change from baseline analyses, used a mixed model for repeated measures, where applicable.
Results
Demographic and baseline clinical characteristics
Overall, this study included 68 participants (36 participants in Cohort 1 and 32 participants in Cohort 2), with 65 participants (96%) completing and 3 participants discontinuing the study treatment (Supplementary Fig. S3). In the insulin-naïve subgroup of Cohort 1, two participants discontinued the study due to a scheduling conflict and in the insulin-naïve subgroup of Cohort 2, one participant was lost to follow-up. Demographics and baseline characteristics were generally similar between the two cohorts (Table 1).
Participant Demographics and Baseline Characteristics
HbA1c, glycated hemoglobin; SD, standard deviation; T2D, type 2 diabetes.
Primary outcome measure
In Cohort 1, required overrides of the CDS dosing recommendation occurred for 2 of 267 injections (0.7%) for the basal-switch subgroup and 3 of 296 injections (1.0%) for the insulin-naïve subgroup. Based on the outcomes in Cohort 1, the CDS algorithm was modified for Cohort 2, which were key modifications to the CDS. In Cohort 2, required overrides of the CDS dosing recommendation occurred for 3 of 240 injections (1.3%) for the insulin-naïve subgroup. No dosing overrides occurred in the basal-switch subgroup of Cohort 2. A summary of the total number of participants by number of overrides and reason for overrides is provided in Table 2 and Supplementary Table S2. All dose overrides were attributed to investigator concerns about the risk of hypoglycemia, and all were deemed medically appropriate.
Number of Participants with Overrides of Dose Recommendation
Secondary and exploratory outcomes
A significant (P < 0.05) reduction in HbA1c from baseline to end of treatment at Week 16 was observed for both the basal-switch and insulin-naïve subgroups in both cohorts (Fig. 2). For Cohort 1, 29.4% of basal-switch participants and 52.6% of insulin-naïve participants achieved HbA1c <7.0% and for Cohort 2, 12.5% of basal-switch participants and 46.7% of insulin-naïve participants achieved HbA1c <7.0% at Week 16. For both cohorts, mean FBG decreased into the 80–120 mg/dL range shortly after efsitora initiation and remained in that range for the rest of the treatment period (Fig. 3). The insulin-naïve population experienced a greater decrease in mean FBG than the basal-switch population in both cohorts. The proportion of participants with FBG targets of 80–100 mg/dL and 80–120 mg/dL increased in both subgroups for both cohorts at Week 16 compared with baseline (Fig. 4). The mean percent time in range of 70–180 mg/dL (3.9–10.0 mmol/L), as measured by CGM, during the final 2 weeks of treatment (Weeks 15–16) was 58.9% and 65.1% for basal-switch and insulin-naïve participants, respectively, for Cohort 1, and 47.3% and 61.7% for basal-switch and insulin-naïve participants, respectively, for Cohort 2 (Fig. 5).

Change in HbA1c from baseline to end of treatment. *P < 0.05; **P < 0.01 versus baseline. Data are presented as mean (SD). HbA1c, glycated hemoglobin; SD, standard deviation.
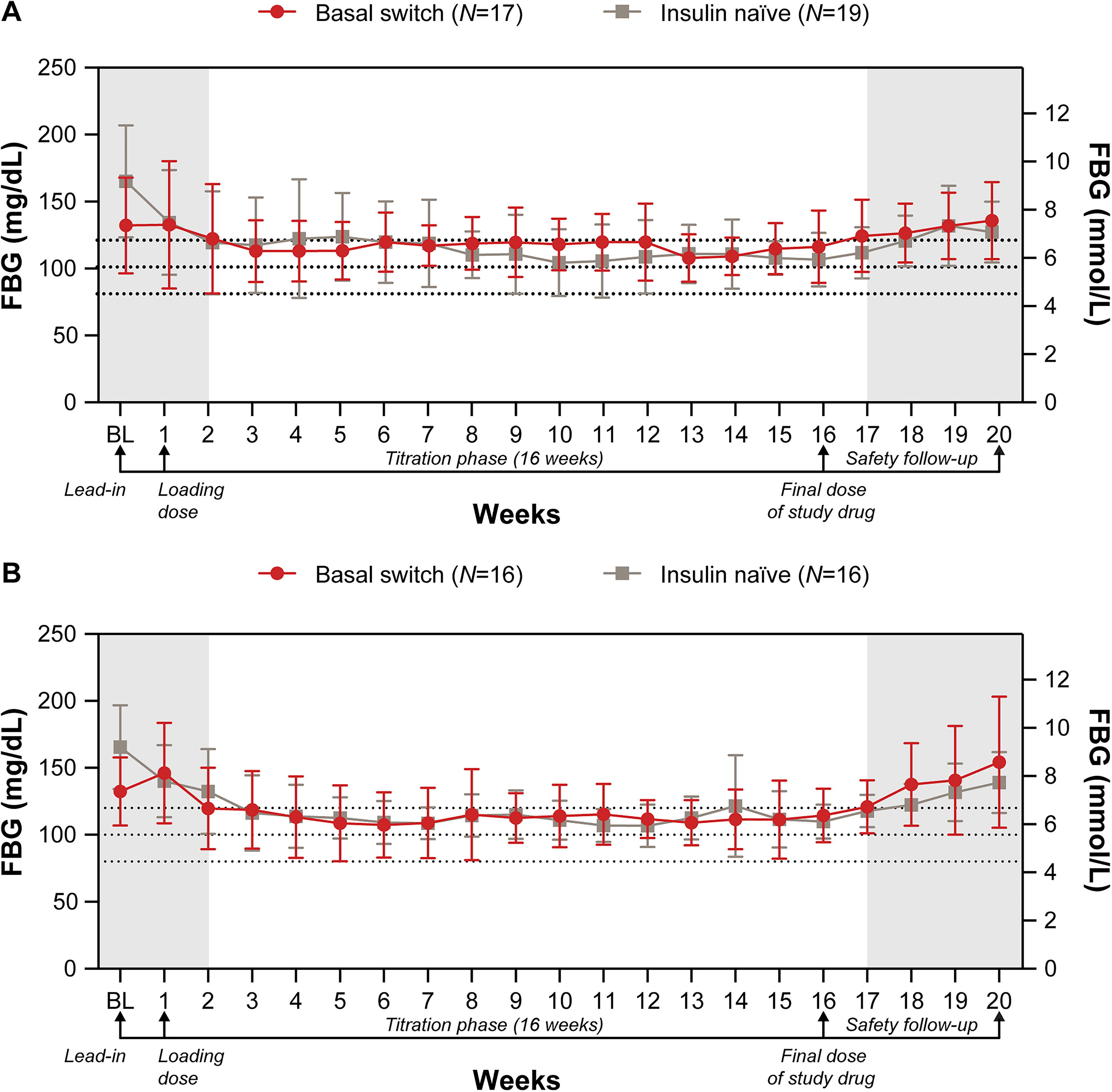
Change in FBG over time for Cohort 1
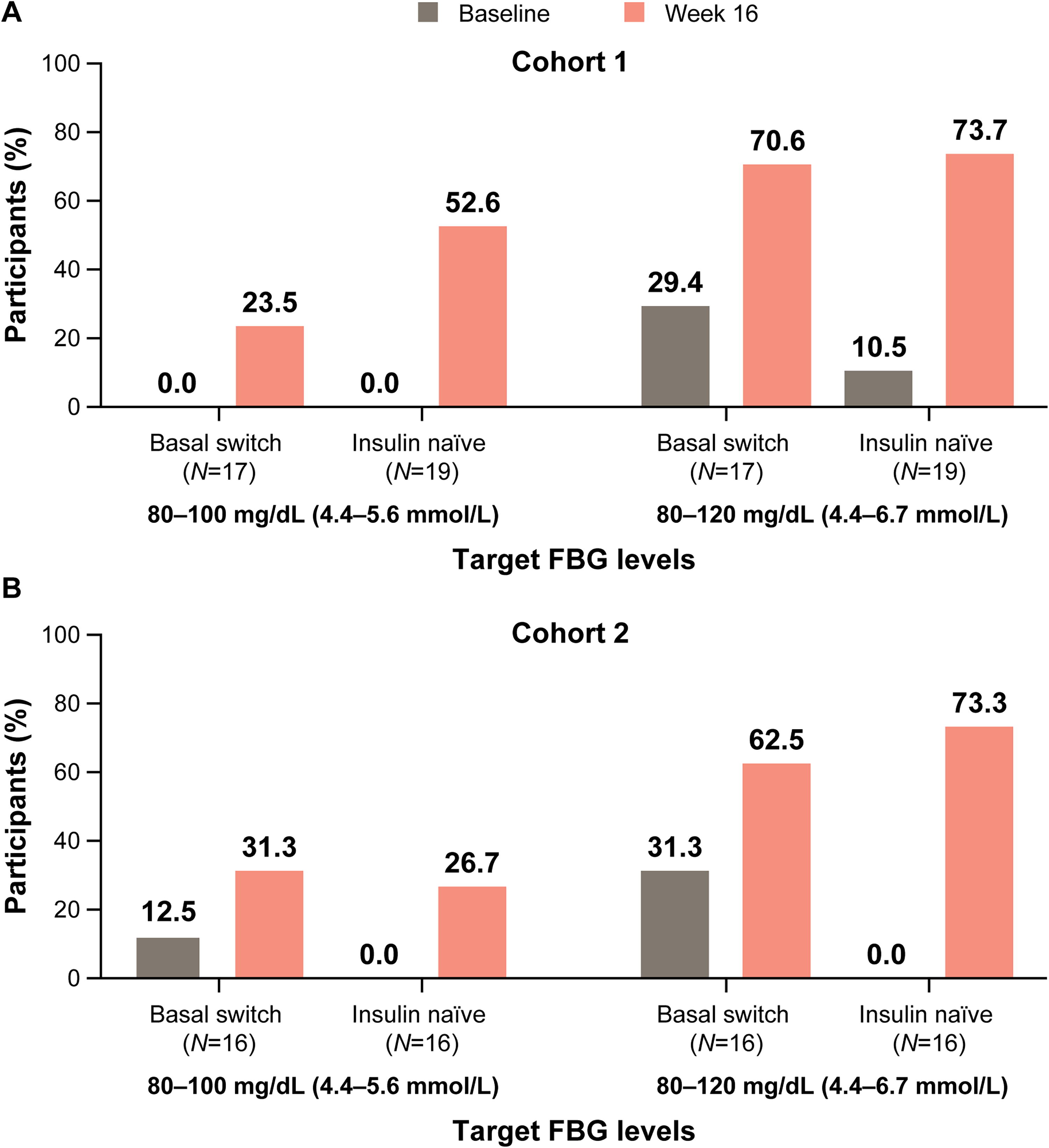
Proportion of participants reaching target FBG at end of treatment for Cohort 1

CGM time in range (24 h) during final 2 weeks of treatment for Cohort 1
Hypoglycemia was analyzed using self-reported glucose data obtained via a study-provided BGM or by CGM. There was only one patient-reported nocturnal hypoglycemic event during the treatment period (data not shown). For hypoglycemic events captured by self-reported glucose monitoring during the treatment period (Weeks 1–16), level 1 hypoglycemic events (<70 mg/dL [3.9 mmol/L]) were more common in the first 4 weeks of the study in Cohort 1 (data not shown). Level 1 hypoglycemic event rates were slightly higher for basal-switch participants in Cohort 1 (12.1 events per participant-year) versus Cohort 2 (7.9 events per participant-year) and were comparable between insulin-naïve participants in Cohorts 1 and 2 (8.1 vs. 8.6 events per participant-year, respectively). Level 2 hypoglycemic (<54 mg/dL [3.0 mmol/L]) event rates were comparable between insulin-naïve participants in Cohorts 1 and 2 (0.3 vs. 0.4 events per participant-year, respectively). Only basal-switch participants in Cohort 1 had reported level 2 hypoglycemia with 1.0 event per participant-year (Table 3).
Hypoglycemia Recorded by Self-Monitoring Blood Glucose and Blinded Continuous Glucose Monitoring
CGM, continuous glucose monitoring; L, level; pt, participant.
For combined level 1 or 2 hypoglycemic events detected using blinded CGM data during the treatment period (Weeks 1–16), basal-switch participants in Cohort 1 had a higher rate (84.3 events per participant-year) versus basal-switch participants in Cohort 2 (38.6 events per participant-year) (Table 3). Higher rates of combined level 1 or 2 hypoglycemic events were reported for insulin-naïve participants in Cohort 1 (82.5 events per participant-year) compared with insulin-naïve participants in Cohort 2 (43.9 events per participant-year). Higher rates of level 2 hypoglycemic events were reported for basal-switch participants in Cohort 1 versus Cohort 2 (18.6 vs. 10.5 events per participant-year, respectively) and insulin-naïve participants in Cohort 1 versus Cohort 2 (22.4 vs. 8.8 events per participant-year, respectively).
Safety
Serious AEs (SAEs) were reported for two participants in the basal-switch subgroup (osteomyelitis and hypertension) and one participant in the insulin-naïve subgroup (humerus fractures/injury) in Cohort 1 (data not shown). For Cohort 2, no SAEs were reported in the basal-switch subgroup and one SAE was reported for the insulin-naïve subgroup (acute respiratory failure). All reported SAEs were deemed unrelated to the CDS. No level 3 hypoglycemic events were reported for either cohort. No deaths occurred during the study.
Overall, four device complaints were recorded, which were related to electronic malfunction or the technical design of the CDS. None of the complaints were related to AEs, and none were classified as an unanticipated adverse device effect.
Discussion
This is the first study to explore the feasibility of using a novel dosing algorithm software system (CDS) utilizing weekly efsitora treatment. Overall, the CDS dosing strategy showed promising clinical performance and favorable investigator confidence as determined by the low rate of dose overrides, all of which were attributed to investigator concerns of hypoglycemia. Furthermore, a low number of AEs were reported and none of the AEs were classified as unanticipated adverse device effects.
In this study, the CDS dosing strategy led to improved glycemic control (lower HbA1c and FBG) relative to baseline, without excess hypoglycemia by self-monitoring blood glucose or excess time below range by CGM. A significant reduction in HbA1c from baseline to end of treatment was observed for both basal-switch and insulin-naïve subgroups in both cohorts. Furthermore, the mean FBG target was reached within 3 weeks of efsitora initiation and was sustained for the remainder of the treatment period for both cohorts. Mean time in range 70–180 mg/dL also increased during the final 2 weeks of treatment for both subgroups in both cohorts. These results indicate that the CDS was successful in recommending doses to improve and maintain glycemic control in both basal-switch and insulin-naïve participants. Recent results from the Phase 3 clinical trial program also indicate strong glycemic efficacy when efsitora has been titrated via a traditional algorithm rather than utilizing CDS. 5
Tight glycemic control was achieved with overall low rates of level 2 hypoglycemia from self-reported blood glucose data of less than one event per participant-year. No severe hypoglycemia was reported for either cohort, suggesting that the CDS was successful in avoiding excess hypoglycemia. Hypoglycemic events detected by blinded CGM were greater than self-reported events. A CDS that utilizes CGM may therefore more effectively identify and react to more episodes of hypoglycemia than a system that uses self-monitoring blood glucose.
Several insulin dosing algorithms have been developed and are undergoing investigation in people with T2D. These include the Diabetes Pal smartphone app, which suggests insulin doses based on FBG data, 12 the Diabetes Insulin Guidance System (DIGSTM) software, which automatically adjusts a patient’s insulin dosage between clinic appointments, 13 the iSAGE RX mobile app that automatically manages the insulin titration plan and provides education on insulin administration, 14 and the My Dose Coach app with web portal that allows health care professionals to define an individualized dose plan, which the app uses to provide dose recommendations based on the user’s FBG and event data. 15 For the DIGS software, 99.8% of insulin dosage adjustment recommendations were approved by the study team. 13 Furthermore, although the number of overrides is not provided, participants using the iSAGE RX mobile app required almost no unexpected visits or phone consultations to manage their insulin doses, suggesting that dose recommendations were accepted. Similarly, in participants with type 1 diabetes who used an artificial intelligence-based decision-support system (The DreaMed Advisor Pro; DreaMed Diabetes Ltd., Petah Tikva, Israel) combined with an insulin pump, the overall percentage of overrides was also low at 1.9%. 16 A limitation of comparing the dosing algorithms or software described above is the difference in how a target or primary outcome was defined in each study. 17 Therefore, the effect cannot be compared across studies or directly compared with the CDS in our study.
Diabetes management tasks, such as blood glucose monitoring, daily insulin injections, and insulin titration decisions, can increase diabetes burden and lead to poor treatment outcomes. 3,18,19 The development of once-weekly insulin receptor agonists has the potential to increase willingness and improve compliance with an insulin therapy in people with T2D. Utilizing a CDS for insulin titration can reduce burden by providing convenient, individualized dose titration support. CDS systems are becoming a critical tool for care and disease management, 20 and there is increasing evidence of the benefits of CDS systems on clinical and economic outcomes. 21 –23
This exploratory study has certain strengths and limitations. The short study duration of 16 weeks, no comparator group to evaluate the study outcomes, and small sample size do not allow the assessment of clinical benefits or substantial safety of the CDS system. Furthermore, close clinical supervision with dosing at a research site and investigator review of hypoglycemic events and tagged FBG are not representative of anticipated real-world use but were appropriate for an early feasibility study. Iterations to allow the algorithm to operate with less oversight are required for use in real-world settings. In addition, CGM input is becoming increasingly popular and would be a natural extension for further CDS development; however, for this first effort only BGM was used, which limits generalizability to patients who are willing to utilize BGM. A CDS that utilizes CGM may more effectively identify and react to episodes of hypoglycemia than a system that uses self-monitoring blood glucose. Although the algorithm could use alternative input to fasting BGM and hypoglycemia, additional tuning of the algorithm would be needed to use CGM input due to the increased hypoglycemia detection with CGM, among other differences. The observed clinical outcomes support a long-term clinical study of the CDS system in an outpatient environment.
Conclusions
In this study, the CDS provided clinically acceptable efsitora dosing recommendations in both insulin-naïve and basal-switch participants. This study provided initial data to refine the CDS, which potentially offers clinical benefits in glycemic outcomes and reduced treatment burden for people with T2D being treated with efsitora. The current CDS was specifically designed for patients on basal insulin alone. It should be noted that the core feedback control approach in the current CDS can be extended for people who also use prandial insulin with design changes for this patient population. The current study provides a basis for the future development of patient-facing decision support incorporating PK/PD data and additional information for personalization of insulin management. Efsitora has been successfully titrated in both standardized, paper-based approaches in the Phase 3 clinical trial program as well as via this CDS algorithm and has shown strong glycemic efficacy across both approaches.
Footnotes
Acknowledgments
The authors would like to thank all the study participants. The authors would also like to thank Bo Zhang, Chris Kovalchick, Donald Jennings, Ming Zhong, Sarah Valentine, Sharon Weigand, and Sylvia Shenouda for their input on the design and implementation of the CDS. Medical writing assistance was provided by Joanna Best, PhD, and Renee E. Granger, PhD, of ProScribe—Envision Pharma Group, and was funded by Eli Lilly and Company. ProScribe’s services complied with the international guidelines for Good Publication practice.
Authors’ Contributions
M.K.: Conceptualization (lead), writing—original draft (lead), and writing—review and editing (equal). C.K.: Writing—review and editing (equal). J.X.: Writing—review and editing (equal) and formal analysis (equal). J.F.: Investigation (supporting) and writing—review and editing (equal). R.B.: Writing—review and editing (equal). J.M.: Writing—review and editing (equal) and formal analysis (equal). S.G.: Project administration (lead), resources (lead), software (lead), and writing—review and editing (equal). E.D.: Conceptualization (supporting), funding acquisition (lead), investigation (lead), software (supporting), supervision (lead), writing—original draft (supporting), and writing—review and editing (equal).
Data Sharing
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the United States and the European Union and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at ![]() .
.
Role of the Sponsor
Eli Lilly and Company was involved in the study design, data collection, data analysis, and preparation of the article.
Author Disclosure Statement
M.K., C.K., J.X., J.M., S.G., and E.D. are employees and shareholders of Eli Lilly and Company. J.F. has received research support from 89bio, Akero Therapeutics, Altimmune, Boehringer Ingelheim, Eli Lilly and Company, Intercept Pharmaceuticals, Ionis Pharmaceuticals, Janssen, Madrigal Pharmaceuticals, Merck, NorthSea Therapeutics, Novartis, Novo Nordisk, Oramed, Pfizer, Sanofi, and Shionogi; has been on advisory boards or consulted for 89bio, Akero Therapeutics, Altimmune, Biomea Fusion, Boehringer Ingelheim, Carnot Therapeutics, Echosens, Eli Lilly and Company, Gilead Sciences, Intercept Pharmaceuticals, Merck, Novo Nordisk, Pfizer, and Sanofi; has been on speakers bureau for Eli Lilly and Company; and is an employee and stockholder of Biomea Fusion, Inc. R.B. has received funding from Eli Lilly and Company for conducting this study and has no other significant conflicts of interest.
Funding Information
This study was funded by Eli Lilly and Company, manufacturer of efsitora.
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.