
Abstract
The potential association of variant surface glycoprotein (VSG) gene expression with clonal expression of virulence in African trypanosomes was addressed. Two populations of clonally related trypanosomes, which differ dramatically in virulence for the infected host, but display the same apparent VSG surface coat phenotype, were characterized with respect to the VSG genes expressed as well as the chromosome telomeric expression sites (ES) utilized for VSG gene transcription. The VSG gene sequences expressed by clones LouTat 1 and LouTat 1A of Trypanosoma brucei rhodesiense were identical, and gene expression in both clones occurred precisely by the same gene conversion events (duplication and transposition), which generated an expression-linked copy (ELC) of the VSG gene. The ELC was present on the same genomic restriction fragments in both populations and resided in the telomere of a 330-kb chromosome; a single basic copy of the LouTat 1/1A VSG gene, present in all variants of the LouTat 1 serodeme, was located at an internal site of a 1.5-Mb chromosome. Restriction endonuclease mapping of the ES telomere revealed that the VSG ELC of clones LouTat 1 and 1A resides in the same site. Therefore, these findings provide evidence that the VSG gene ES and, potentially, any cotranscribed ES-associated genes do not play a role in the clonal regulation of virulence because trypanosome clones LouTat 1 and 1A, which differ markedly in their virulence properties, both express identical VSG genes from the same chromosome telomeric ES.
Introduction
The different biological traits noted above can be defined as primary virulence factors for all African trypanosomes because parasites in which the relevant genes have been deleted, mutated, or knocked down are unable to infect or survive in specific mammalian hosts (Leal et al., 2001; Hoek et al., 2002; Wang et al., 2003). A primary focus of this study, however, has been on relative virulence of trypanosomes for their mammalian hosts. It is well known that different isolates, species, subspecies, and laboratory lines of trypanosomes exhibit remarkable variation in pathogenicity and virulence for genetically defined host species (McNeillage and Herbert, 1968; Mulligan, 1970; Clayton, 1978; Barry et al., 1979; Inverso and Mansfield, 1983). The underlying question has been whether such biological differences are immutable characteristics associated with genetically distinct populations of trypanosomes, or whether intraclonal biological variation occurs in trypanosomes, which impacts on the course of infection within an individual animal host.
This question was addressed in an earlier study from our laboratory in which inbred mice of a relatively resistant phenotype (Levine and Mansfield, 1981; DeGee and Mansfield, 1984; De Gee et al., 1985, 1988) were each infected with a single T. brucei rhodesiense LouTat 1 trypanosome (Inverso and Mansfield, 1983; Inverso et al., 1988). Several different variant antigenic types (VATs) were isolated from parasitemia peaks at intermediate and late time points during the ensuing infection; these VATs were subcloned and characterized as to VSG phenotype. Three different VATs, which represented antigenically distinct daughter cell populations clonally derived from a single LouTat 1 parental cell in a single mouse, were then used to infect resistant mice; the progression of infection was monitored in comparison with mice infected with the parental cell LouTat 1. The surprising result was that each daughter cell population exhibited a different virulence phenotype compared with the parental clone. For example, LouTat 1 caused death at approximately 62 days postinfection, whereas LouTat 1.3, 1.4, and 1.5 caused death at approximately 44, 30, and 28 days, respectively. These results demonstrated that VATs arising during infection initiated by a single cell expressed virulence phenotypes different from the infecting VAT. In essence, daughter cells arising within a trypanosome population expressed the capacity to transcend host genetic resistance characteristics and render a relatively resistant animal into a more susceptible one. As virulence expression appears to be a progressive phenotype during chronic trypanosome infections, this clonal characteristic recently has been termed a “virulence rheostat” (Mansfield, 2006).
Given that different VATs exhibited differences in virulence, and that other biological traits of trypanosomes may be controlled by genes with VSG-like characteristics (e.g., SRA) or by ESAGs expressed within an active VSG gene ES (Borst et al., 1998; Hoek et al., 2000, 2002; Hoek and Cross, 2001; Berriman et al., 2002), a logical question was whether the VSG gene, or the ES used by specific VSG genes, in virulent trypanosomes plays a role in relative virulence. Therefore, we initially hypothesized that trypanosome virulence may be determined directly by the VSG molecule itself. Such an hypothesis has an experimental basis because VSG molecules and their substituents have been shown to express or induce biological activities independent of their status as variant antigens (Musoke and Barbet, 1977; Mathias et al., 1990; Lucas et al., 1994; Magez et al., 1997, 1998; Coller et al., 2003; Leppert et al., 2007; Lopez et al., 2008). Alternatively, we proposed that the expression of virulence may be linked to the ES utilized by VSG genes. The rationale was that transcription of certain VSG genes may include cotranscription of hypothetical virulence regulatory genes, if such genes are ESAGs unique to a specific VSG ES.
We subsequently derived and characterized a subclone of LouTat 1, designated clone LouTat 1A, which was shown to be highly virulent for mice (Inverso et al., 1988). Immunological and biochemical examinations of these organisms showed that T. brucei rhodesiense LouTat 1 and 1A both express a similar surface coat phenotype (Inverso et al., 1988; Reinitz et al., 1992). These findings suggested, but did not prove, that virulence is not associated with the VSG molecule. In this article, therefore, we address the hypothesis in greater detail, that is, the potential linkage of the expressed VSG gene or its ES with virulence regulation in LouTat 1 and LouTat 1A populations of T. brucei rhodesiense. To this end, VSG cDNA was sequenced from both organisms, and VSG probes were used to characterize the mechanism of LouTat 1/1A VSG gene expression, the genomic organization of this VSG gene, and the ES employed in members of the T. brucei rhodesiense LouTat 1 serodeme. Using these approaches, we determined that virulence expression in these populations was independent of both the VSG gene and the telomeric site from which the VSG gene is transcribed.
Materials and Methods
Trypanosomes
The isolation of T. brucei rhodesiense clone LouTat 1 and the derivation of antigenic variants 1.3, 1.4, and 1.5 have been described previously (Levine and Mansfield, 1981; Inverso and Mansfield, 1983; Inverso et al., 1988). The highly virulent LouTat 1A subclone was obtained by serial subpassage of the less-virulent LouTat 1 clone through immunocompromised mice, as we have reported (Inverso et al., 1988). Trypanosomes used as sources of RNA and DNA were grown by infection in immunosuppressed mice (cyclophosphamide treated; 300 mg/kg) (Smith et al., 1982) with the relevant trypanosome stabilates. Mice were exsanguinated at 3–4 days postinfection and trypanosomes separated from blood by DEAE cellulose column chromatography (Lanham and Godfrey, 1970). Purified parasites were washed two times at 4°C with 10 mM phosphate-buffered saline (pH 8.0) containing 1% glucose. After the final wash, parasites were stored as frozen pellets in liquid nitrogen until use.
cDNA amplification and VSG cloning procedure
Cloning of full-length VSG cDNA was accomplished by methods previously described (Reinitz et al., 1992). Polyadenylated RNA was isolated from frozen trypanosome pellets by conventional procedures of polytron homogenization in 8 M guanidinium hydrochloric acid (HCl), centrifugation through 5.7 M CsCl cushions, and poly(A+) RNA selection on oligo dT columns. RNA was reverse transcribed using 200 units moloney murine leukemia virus (M-MLV) reverse transcriptase and a 28-bp oligonucleotide primer based on a 3′ conserved sequence present in all VSG transcripts (Merritt et al., 1983) (GGGTAACTTACGTGTTAAAATATATCAG). The following reaction conditions were used: 10 μg polyA RNA, 0.5 μg 28mer, 500 μM dNTPs, 2 μg BSA, 40 units RNAsin, 1 μg actinomycin D, and 0.4 μg globin RNA in 10 mM Tris [pH 8.3], 0.15 mM KCl, 0.10 mM DTT, and 3 mM MgCl2. The 50 μL reaction mixture was incubated at 37°C for 60 min, then 31 μL of 150 mM NaOH was added and incubated at 65°C for 1 h. The pH was adjusted to 8.0 and the DNA precipitated by adding 31 μL of 1 M Tris, 31 μL of 1 M HCl, 15 μL of 3 M NaOAc, and 388 μL ethanol. The chelating agent ethylenediaminetetraacetic acid (EDTA) was not added because of interference with Mg standardization in the subsequent PCRs.
First strand cDNA was resuspended in 60 μL water and 5 μL was added to PCRs containing 3 μM of each dNTP, 5 mM MgCl2, 1 μM of each oligonucleotide (3′ 28mer above, and a 5′ 26mer [CGCTATTATTAGAACAGTTTCTGTAC]), derived from the trypanosome mini-exon-derived spliced leader sequence (Cross, 1990; Cross et al., 1990), 2.5 units Amplitaq™ DNA polymerase (Perkin Elmer, Waltham, MA) in 10 mM Tris (pH 8.3), and 50 mM KCl in a total volume of 100 μL. Reaction mixtures were overlaid with 100 μL mineral oil, heated to 97°C for 5 min, slowly cooled to 45°C, and subjected to 30 cycles of amplification (MJR Thermal Cycler) with the following profile: 93°C for 1 min, 66.5°C for 1 min, and 72°C for 3 min. Samples were then concentrated 10-fold by ethanol precipitation and electrophoresed through 1.3% Sea Plaque GTG™ agarose (FMC Corp., Philadelphia, PA), and the full-length 1732-bp cDNA band was isolated by phenol extraction. This DNA was then ethanol precipitated and resuspended in 10 μL of 1 × Klenow buffer (50 mM Tris [pH 7.7], 10 mM MgCl2, 1 mM DTT, 50 μg/mL BSA, 42 mM dNTPs) and 5 units of the Klenow fragment of DNA polymerase I. The reaction was incubated at 30°C for 30 min, 75°C for 10 min, cooled to 4°C prior to adding 4 μL of 5 × T4 polynucleotide kinase buffer (350 mM Tris [pH 8.0], 50 mM MgCl2, 25 mM DTT), 1 μL of 10 mM ATP, 10 units polynucleotide kinase, and 13 μL water to give a final volume of 50 μL. Kinasing reactions were carried out by incubating for 1 h at 37°C and 10 min at 75°C, adjusted to 400 μL and 300 mM sodium acetate, extracted with phenol:chloroform:isoamyl alcohol (25:24:1), and ethanol precipitated. This was resuspended in 20 μL ligation mix containing 10 ng SmaI-restricted pBluescript II KS + ™ (Stratagene), 8 units T4 DNA ligase (no. 600011; Stratagene), 500 μM ATP, 50 mM Tris (pH 8.0), 7 mM MgCl2, and 1 mM DTT and was incubated overnight at 13°C. An aliquot of the ligation reaction (0.3 μL) was diluted 10-fold and used to transform competent Escherichia coli (strain DH5α F′), which were then selected by growth on Luria broth agar plates containing ampicillin (100 μg/mL), isopropyl beta-
DNA sequencing strategy
The VSG cDNA was sequenced primarily as a double-stranded (ds) template as originally described by Sanger (Sanger et al., 1977; Sambrook et al., 1989); however, certain regions were sequenced using single-stranded (ss) DNA. By a combination of these methods, both strands were completely sequenced. All reactions used the Sequenase™ enzyme and conditions were as described by the manufacturer (U.S. Biochemical, Cleveland, OH). A series of eight oligonucleotides (16–22 bp) were synthesized on an Applied Biosystems (Foster City, CA) 380B DNA synthesizer for use as primers in combination with various templates (as described below). Four oligonucleotides primed each strand. Their sequences and corresponding VSG gene positions (Fig. 1) were as follows: (1) AACAGCTATGACCATG, plasmid polylinker; (2) GGACGCAAGTCTACCTAGCAGC, 336–357; (3) TAAACATAGCGCACGAGACC, 519–536; (4) GACAGCAGAGCTAACAACAG, 991–1010;
and on the complementary strand, (5) GTAAAACGAACGGCCAGT, plasmid polylinker; (6) GTGTAACAGGCACACCAC, 1469–1452; (7) CTGCAAGTGTCTACCGATGCC, 1341–1321; (8) GTGGTCTGCGCTTCTGCTAG, 486–467.

Complete nucleotide and deduced amino acid sequence of the variant surface glycoprotein (VSG) cDNA expressed by Trypanosoma brucei rhodesiense clones LouTat 1 and 1A. The precursor VSG molecule is 510 amino acids, which is processed to 461 residues by cleavage of a 26-amino acid hydrophobic leader and a 23-amino acid C-terminal extension (underlined). The 187-bp cDNA fragment derived from pTbr-VSG1a-4 as a probe for northern and selected Southern analyses is denoted by dotted underline. The 484-bp fragment generated by PCR for use as a probe in Southern analyses is denoted by solid underline within the DNA sequence. The accession number for the LouTat 1 VSG sequence is X56643 (Reinitz et al., 1992).

The ds templates included cloned ExoIII deletions and BssHII restriction fragments of the VSG cDNA, whereas the ss sequences were linear fragments obtained using selected pairs of the primers in asymmetric PCRs. The cDNA was found to possess a single BssHII site and the two fragments (742 and 990 bp) were subcloned separately into pBluescript and sequenced using the plasmid polylinker primers 1 and 5. The ExoIII deletions were performed as described by the manufacturer (Stratagene). Briefly, susceptible 5′ and protected 3′ overhangs were created by digestion with EcoRI and KpnI, respectively. Linearized templates were digested with 200 units ExoIII and aliquots were removed every 30 s, frozen, and subsequently treated with S1 nuclease and Klenow prior to religation and transformation back into E. coli DH5α. Such techniques resulted in the isolation of two useful subclones representing nucleotides 649–1732 and 1180–1732, which were sequenced in both directions using primers 1 and 5. Generation of ss templates was accomplished using oligonucleotides 1 and 7 (1:50) with the ExoIII deletion fragment (649–1732) as a template in the asymmetric PCR under the following conditions: 10 ng DNA, 20 nM oligo 1, 1 mM oligo 7, 5 mM MgCl2, 10 mM Tris (pH 8.3), 50 mM KCl, 2.5 units Ampitaq, and 300 μM dNTPs. The 100 μL reactions were subjected to 30 cycles of 95°C for 1 min, 31°C for 30 s, and ramped over 90 s to 72°C for 1 min. Following amplification, ss cDNA templates were precipitated, resuspended in water, passed over CL-6B Sepharose™ spin columns, and sequenced as described (U.S. Biochemical) with primer 7.
Preparation of LouTat 1/1A VSG cDNA probes
A 395-bp VSG cDNA fragment, contained in the recombinant pGEM-3Z plasmid pTbrVSGA-4 (Reinitz et al., 1992), was isolated by double digestion with BamHI and EcoRI, purified from 1.2% low-melting-temperature agarose gels, and digested with HpaII to generate a 187-bp sequence representing an internal, variant specific sequence within the cDNA (Fig. 1). This fragment was further purified from 1.5% low-melting-temperature agarose gels and used as a probe for all subsequent northern blot analyses. The probe used in Southern blots was a 484-bp PCR fragment, which was generated using oligonucleotides annealing to nucleotides 336–357 (described in the sequencing strategy) and 800–819 (ACCTTGCTGTCTTCGGTGTT) of the VSG DNA sequence shown in Figure 1. The probe was purified from the PCR reactants by EtOH precipitation. 32P-labeled cDNA probes were prepared to high specific activity (>1 × 108 cpm/μg) by a standard nick translation protocol (Sambrook et al., 1989) or by direct incorporation of α-32P-dCTP in the PCR amplification reaction.
Northern blot analysis
Total RNA (5 μg/lane) was fractionated in 1.5% agarose gels containing 2.2 M formaldehyde (Lehrach et al., 1977; Sambrook et al., 1989). Following electrophoresis, gels were stained in ethidium bromide (0.5 μg/mL water), destained in dH2O, soaked in 10 × saline/sodium citrate (SSC) for 30–45 min to remove formaldehyde, and transferred to GeneScreen Plus (DuPont, Boston, MA) via capillary blotting in 10 × SSC (1.5 M NaCl, 0.15 M sodium citrate, pH 7.0) for 16–20 h. After transfer was complete, membranes were baked at 80°C for 1.5 h. Hybridization and posthybridization washes were performed as stated (see figure legends).
Southern blot analysis
Genomic DNA was prepared from frozen cell pellets as described elsewhere (Milhausen et al., 1983). Restriction enzyme digests of genomic DNA were performed by addition of 25 units restriction endonuclease to 10 μg DNA in 25 μL of 1 × buffer provided by the supplier (BRL, Gaithersburg, MD). Digestion was carried out for 4 h at 37°C. The entire reaction mixture was loaded into a well of an agarose gel for electrophoresis in 1 × TBE (tris/borate/EDTA buffer, pH 8.0) (89 mM Tris, 89 mM boric acid, 2 mM EDTA). Fractionated DNA was visualized by ethidium bromide staining (0.5 μg/mL in water). Prior to transfer the DNA was partially depurinated (0.25 M HCl for 15 min) and denatured (30 min in 0.4 M NaOH–0.6 M NaCl). Gels were neutralized by soaking in 0.5 M Tris (pH 7.5) and 1.5 M NaCl for at least 30 min prior to transfer, and DNA was transferred onto nitrocellulose membranes (Schleicher and Schuell, Keene, NH).
Filters containing DNA were prehybridized for 1 h at 42°C in buffer containing 5 × SSPE (saline/sodium phosphate/EDTA buffer, 0.74 M NaCl, 0.05 M NaH2PO4·H2O, 5 mM EDTA), 10 × Denhardt's solution, 0.5% sodium dodecyl sulphate (SDS), and 50% formamide. Heat-denatured, nick-translated probe was added to the hybridization buffer at 107 cpm/mL with 100 μg/mL denatured salmon sperm DNA. The blots were hybridized overnight at 55°C. Posthybridization washes were done at 65°C in 1 × SSC and 0.1% SDS (2 × , 30 min) followed by two washes at 65°C for 30 min each in 0.1 × SSC and 0.01% SDS.
Bal31 exonuclease digestion
Genomic DNA was isolated as described above for Southern blot analysis. Bal31 exonuclease digestions were performed by incubation of 1 unit enzyme/20 μg genomic DNA in digestion buffer (12 mM CaCl2, 12 mM MgCl2, 0.2 M NaCl, 20 mM Tris HCl [pH 8.0], 1 mM EDTA) at 30°C for different periods of time. Reactions were terminated by addition of EDTA (pH 8.0) to a final concentration of 20 mM. Samples were deproteinized immediately by phenol extraction and precipitated with ethanol. DNA recovered from the ethanol precipitation was suspended in the appropriate buffer and digested with excess XmnI (10 units/μg DNA) for 4 h at 37°C. The entire reaction mixture was loaded into a well of a 1.2% agarose gel and electrophoresed, transferred to nitrocellulose, and hybridized as described earlier for Southern blot analysis.
Pulsed-field gradient gel electrophoresis
Samples were prepared by embedding live trypanosomes in agarose blocks and subsequently lysing the embedded cells in proteinase K and SDS as described by Scholler et al. (1986). The pulsed-field gel electrophoresis (PFGE) approach was adapted from the methods of Schwartz and Cantor (1984). Gels containing 1% high-strength agarose (BioRad, Hercules, CA) in 0.5 × TBE were cast in 18 cm × 18 cm glass supports; agarose blocks containing approximately 3.8 × 107 cells were loaded into the wells. Gels were subsequently run in a CHEF II apparatus (BioRad) in 0.5 × TBE running buffer at 200 V and 12°C with equal alternating pulse times of 60 s for approximately 15 h and then for 6 h with a 90 s switch time. Following electrophoresis, the gels were stained with ethidium bromide to visualize chromosomal bands. The gels were transferred and hybridized as described for Southern blot analysis.
Results
Trypanosome clones LouTat 1 and 1A express the same VSG gene
Full-length cDNAs representing the VSG genes expressed in clones LouTat 1 and 1A were derived from poly(A+) RNA of the respective organisms by selective PCR amplification, as previously described (Reinitz et al., 1992). Briefly, a 26-bp oligonucleotide primer corresponding to the 5′ spliced leader sequence (mini-exon-derived sequence) of trypanosome mRNAs and a 28-bp oligonucleotide primer corresponding to a conserved 3′ sequence common to trypanosome VSG transcripts were used to amplify VSG cDNA. The resultant ds cDNA was treated to repair 5′ and 3′ overhangs, and a phosphate was added prior to blunt-ended ligation into a suitable vector. The full-length cDNA encoding LouTat 1 and LouTat 1A VSG molecules was sequenced in both directions, and the nucleotide sequences were shown to be identical for both clones (see Fig. 1 for nucleotide and deduced amino acid sequences). The LouTat 1/1A VSG cDNA encodes a precursor protein of 510 amino acids with a 26-residue N-terminal hydrophobic signal sequence and a C-terminal extension of 23 amino acids. Based on earlier partial amino acid sequence analyses of the VSG proteins from LouTat 1 and 1A (Inverso et al., 1988), we predict that the precursor molecule is processed to a mature VSG of 461 amino acids, beginning at the N-terminus with threonine 27 and ending with conserved residues common to type I VSG molecules at the C-terminus. The LouTat 1/1A VSG molecule also contains conserved residues throughout the molecule, which are common to VSGs as a family (Rice-Ficht et al., 1981; Reinitz et al., 1992; Blum et al., 1993).
To make a variant-specific probe, a recombinant plasmid, pTbrVSGA-4, was generated, which contains a VSG cDNA fragment cloned into the BamHI-EcoRI site of pGEM3Z. Probes for northern blot analyses were generated by digestion of the BamHI-EcoRI purified insert with HpaII to remove the 3′ terminal 208-nucleotide sequence that contained conserved sequences common to VSG genes. The variant specific 5′ 187-bp fragment isolated from this digestion was labeled for use in all subsequent northern blot and selected Southern blot analyses. An additional probe used in some Southern blots was a 484-bp PCR fragment that was generated by direct PCR using oligonucleotides annealing to nucleotides 336–358 (GGACGCAAGTCTACCTAGCAGC) and 800–819 (ACCTTGCTGTCTTCGGTGTT) of the VSG DNA sequence shown in Figure 1.
To confirm that the VSG probe derived from pTbr-VSGA-4 was variant specific, Northern blot analysis was performed. Total RNA from trypanosome populations was size-fractionated by agarose gel electrophoresis, transferred to nylon membranes, and hybridized to the labeled 187-bp VSG cDNA fragment. As shown in Figure 2, the probe hybridized equivalently to RNA from variants LouTat 1 and 1A but not to RNA from the heterologous variant LouTat 1.5 or from T. brucei procyclic forms, which do not express VSG genes. Additionally, the cDNA probe did not hybridize to RNA from other heterologous variants of the LouTat 1 serodeme such as LouTat 1.3 or 1.4 (not shown). The size of the RNA species that hybridized to the VSG cDNA probe was determined to be approximately 1.7 kb, which was a transcript size compatible with the predicted VSG molecule size. A control probe (NVS1a-12) was rehybridized to RNA from all populations, as shown in Figure 2. NVS1a-12 is a cDNA selected from a LouTat 1A cDNA library, which hybridized to an RNA species similar in size and abundance to that recognized by the VSG-specific cDNA probe; it does not display differential hybridization patterns among variants and, as such, represents a nonvariant specific sequence (a similar pattern of control hybridization occurs with conventional probes such as α/β tubulin, as we have shown) (Reinitz et al., 1992; Schopf and Mansfield, 1998; Demick et al., 2010). Overall, therefore, these results support previous immunological and biochemical evidence that LouTat 1 and 1A express the same VSG surface coat, and that this coat is distinct from VSGs expressed by other variants of the LouTat 1 serodeme (Inverso et al., 1988; Reinitz et al., 1992; Schopf et al., 1998).
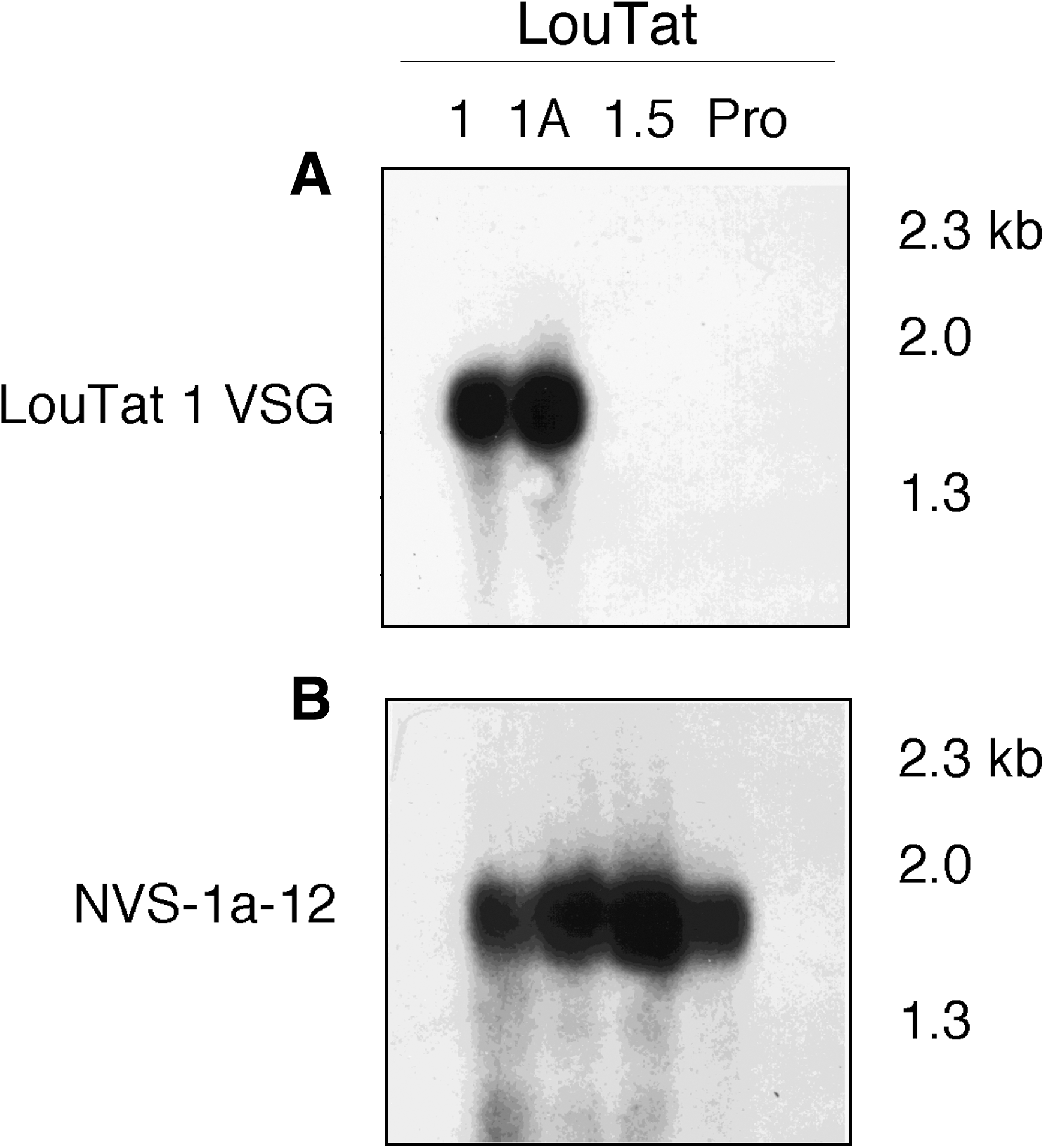
Variant specificity of the LouTat 1 VSG cDNA probe as shown by northern analysis. Five microliters of total RNA from LouTat clones 1, 1A, and 1.5 and T. brucei procyclic forms (Pro) were size fractionated in 1.5% formaldehyde agarose gels, transferred to nylon membranes by capillary blotting, and baked at 80°C for 1.5 h. Filters were prehybridized at 42°C for 1 h in buffer containing 50% formamide, 10% dextran sulfate, l M NaCl, and 1% SDS. To the hybridization buffer was added 100 μg/mL denatured salmon sperm DNA and 1 × 105 cpm/mL of the 32P-labeled 187-bp VSG1a-4 (LouTat 1 VSG) cDNA fragment (
LouTat 1 and 1A express their VSG gene by the same mechanism
The genomic organization of the VSG gene was examined by Southern blot analyses with the 187- and/or 484-bp VSG cDNA probes noted earlier. Hybridization of either probe to genomic DNA from clones LouTat 1, 1A, 1.3, 1.4, and 1.5 restricted by several enzymes is shown in Figure 3. The probes hybridized to two restriction fragments in DNA from expressor population LouTat 1. The smaller band of clone LouTat 1 was approximately 8.7 kb and the larger band was approximately 22 kb in size. Similarly, the probes hybridized to two fragments in genomic DNA from clone LouTat 1A; these were a large fragment of approximately 23 kb and a smaller 7.6 kb band. Nonexpressor populations LouTat 1.4 and 1.5 each contained a single restriction fragment, 3 and 12.2 kb, respectively, which hybridized to these probes. The nonexpressor variant LouTat 1.3 also contained a single band of 10.3 kb, although a weakly hybridizing fragment was apparent directly below this band at approximately 9.8 kb. Because SacI does not cut within the cloned VSG cDNA, the bands detected in the Southern blot described earlier represent discrete copies of the VSG gene.

Southern blot analysis of LouTat 1/1A VSG gene organization in different trypanosome populations shows that there are two gene copies in the expressor cells. Ten micrograms of genomic DNA from LouTat variants 1, 1A, 1.3, 1.4, and 1.5 were digested in 25 μL restriction digestion buffer with 25 units SacI for 4 h at 37°C. Digested DNA was size-fractionated in a 0.55% agarose gel and transferred to nitrocellulose membrane. The blots were prehybridized in a buffer containing 10 × Denhardt's solution and 0.5% SDS, and 5 × SSPE and 0.5% SDS at 42°C for 1 h. Denatured salmon sperm DNA (100 μg/mL) and 32P-labeled 484-bp LouTat 1 VSG PCR-derived cDNA fragment (107 cpm/mL hybridization buffer) were added and the blots incubated at 55°C overnight. Posthybridization washes were performed at 65°C in 1 × SSC–0.1% SDS (2 × , 30 min), followed by two high-stringency washes at 65°C for 30 min each in 0.1 × SSC–0.01% SDS.
Differences in restriction fragment size of the VSG gene copies in expressor clones LouTat 1 and 1A were observed. This is not unexpected because actively expressed VSG genes located in telomeric ES often display size variability due to differences in subtelomeric sequences. However, we also observed differences in restriction fragment size among basic copy (BC) fragments of expressor and nonexpressor variants, suggesting that the BC gene may be located near a telomere. Therefore, we examined for a potential association of both VSG copies in the expressor populations with chromosome telomeres. Genomic DNA from several variants was digested with Bal31 exonuclease, followed by digestion with selected restriction endonucleases. Shown in Figure 4 are the representative results of Bal31 digestion studies with DNA from LouTat 1 and 1A. Genomic DNA from each variant was digested with Bal31 for different periods of time, purified, and subsequently digested with XmnI. DNA was size fractionated in agarose gels, transferred to nitrocellulose, and hybridized with the 484-bp VSG cDNA probe described earlier. Only the lower bands of expressor variants LouTat 1 and 1A displayed relative sensitivity to Bal31 exonuclease digestion (Fig. 4). The upper VSG fragment, presumably the BC, did not display similar sensitivity to Bal31 exonuclease digestion; this was similar to other chromosome interior genes such as α/β tubulin, which were not affected by Bal31 treatment (not shown). Thus, these results suggest that the expression-linked copy (ELC) but not BC of the LouTat 1/1A VSG gene is telomere associated.

Relative Bal31 exonuclease sensitivity of the VSG expression-linked copy (ELC) gene in LouTat 1/1A demonstrates that the ELC but not the basic copy (BC) gene in expressor trypanosomes is located near a chromosome telomere. Twenty micrograms of genomic DNA from LouTat 1 and 1A was digested with 1 unit Bal31 exonuclease at 30°C for different lengths of time. Samples were deproteinized, precipitated with ethanol, and digested with XmnI. DNA was size-fractionated in 0.55% agarose gels, transferred to nitrocellulose, hybridized with a 32P-labeled LouTat 1 VSG cDNA probe, and washed as described in Figure 3.
Trypanosome clones LouTat 1 and 1A express their active VSG gene from the same chromosome telomeric site
We next determined if the expressed VSG ELC in LouTat 1 and 1A was on the same or different restriction fragments. Because of restriction fragment size variability of the VSG bands seen in expressor variants LouTat 1 and 1A as shown in Figure 3, it was difficult to determine the identity of ELC or BC by digestion with SacI or several other restriction enzymes. Therefore, to eliminate restriction fragment size variability related to telomere end sequence variability, we digested genomic DNA from all variants with XmnI, which, based on nucleotide sequence analysis, cuts at a single site within the 3′-region of VSG cDNA near the polyadenylation site. Genomic DNA from LouTat clones 1, 1A, and 1.5 was digested with XmnI, size fractionated by agarose gel electrophoresis, transferred to nitrocellulose, and hybridized with the 484-bp VSG probe (Fig. 5). Two bands were apparent in lanes containing the LouTat 1 or 1A DNA, representing a 2.4-kb ELC and 4.5-kb BC fragment. A single 4.5-kb BC band was apparent in XmnI digests from the nonexpressor variant LouTat 1.5. The presence of a single 4.5 kb band in Southern blots with XmnI-restricted genomic DNA from LouTat 1.3 (data not shown) suggests that the smaller weak band observed in Southern blots of SacI-digested DNA (Fig. 3) was due to telomere heterogeneity in the clonal population and does not represent a lingering ELC. The size of the ELC band in clones LouTat 1 and 1A is the same, and it therefore appears that the size variability previously observed in SacI restriction digests was also due to heterogeneity in telomere size between the two populations because XmnI digestion removes VSG subtelomeric sequences. Thus, these results suggest that the VSG ELC of LouTat 1 and 1A is present on the same restriction fragment and, potentially, within the same telomeric ES.
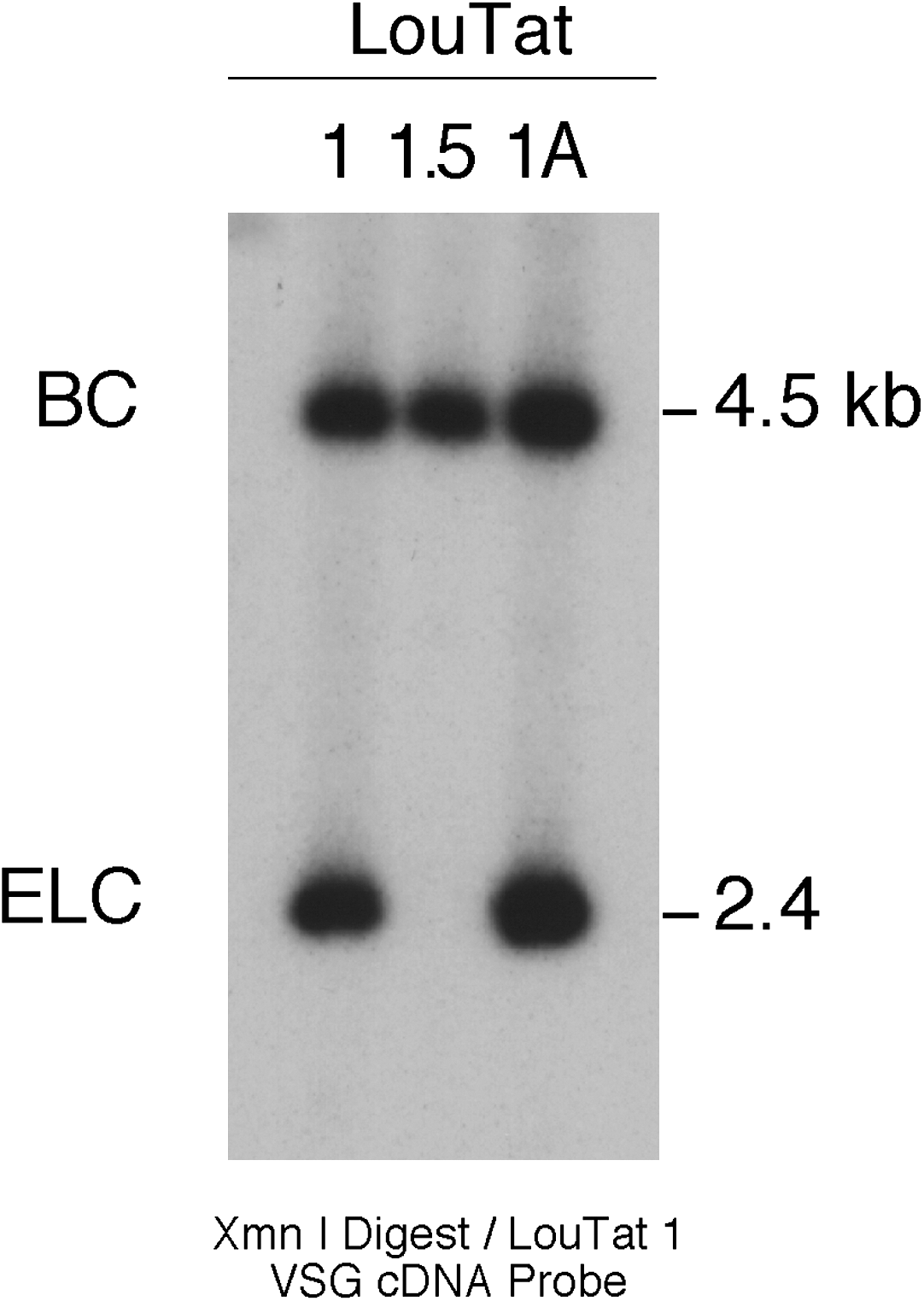
Identification of ELC and BC fragments. Ten micrograms of genomic DNA from LouTat clones 1, 1A, and 1.5 were digested with XmnI, fractionated in a 0.55% agarose gel, transferred to nitrocellulose, hybridized with a 32P-labeled LouTat 1 VSG cDNA probe, and washed as detailed in Figure 3.
To determine whether the VSG ELC of LouTat 1 and 1A is expressed from the same chromosome, PFGE followed by Southern blot analysis was performed. LouTat 1A, the highly virulent trypanosome population, displays a chromosomal pattern identical to the VSG homologous but less virulent LouTat 1 and the heterologous variant LouTat 1.5. The chromosomal patterns are shown in ethidium bromide-stained gels (Fig. 6). When DNA in this gel was transferred to nitrocellulose and hybridized with a VSG cDNA probe, it was observed that the VSG BC gene, present in all VATs, is located on a chromosome of approximately 1.5 Mb (Fig. 6). The ELC of LouTat 1 and 1A was located on an intermediate-sized chromosome of approximately 330 kb, which appears to be the same in both populations. Thus, results of both Bal31 digestion and PFGE analysis indicate that the ELC of the LouTat 1 and 1A VSG gene is expressed from a telomeric ES located on an intermediate-sized chromosome, whereas the BC of the VSG gene resides in the interior of a larger chromosome.

Chromosomal location of LouTat 1/1A VSG ELC genes. Blocks containing approximately 3.8 × 107 trypanosomes from LouTat 1, 1A, and 1.5 were embedded in a 1% high-strength agarose gel and electrophoresed in 0.5 × TBE for 15 h at 200 V and 12°C with equal alternating pulses of 60 s, followed by 6 h with a 90 s switch time. The gel was transferred to a nylon membrane, hybridized with a 32P-labeled LouTat 1 VSG cDNA probe, and washed as described in Figure 3. Bands seen at the very top of the blot are from wells loaded with trypanosomes embedded in agarose blocks.
The foregoing restriction enzyme digestion studies and PFGE analysis demonstrated that the VSG gene ELC of LouTat 1 and 1A resides on a similar sized chromosome. However, more definitive evidence that both variants utilize the same ES is presented in Figure 7. Restriction mapping analysis of LouTat 1 and 1A ELC and BC genes was performed by standard restriction endonuclease digestion analyses combined with Southern blot analyses using the 5′ VSG 484-bp cDNA probe. Results presented here demonstrate that the restriction map of the VSG gene and sequences 5′ to the VSG gene are the same for LouTat 1 and 1A ELC. The upstream barren region of the ES, devoid of restriction sites, was determined to be approximately 10–15 kb. Additionally, the restriction map 5′ to the upstream barren region was determined by digestion studies using eight different restriction endonucleases that cut in this region. No differences in restriction sites in this region were apparent between trypanosomes LouTat 1 and 1A. The BC restriction map of LouTat 1A, which is the same for LouTat 1 and 1.5, differs from the ELC map in regions immediately upstream of the XmnI site 5′ to the VSG gene. The barren region of the BC gene appears to be further upstream than the barren region of the ELC, and it could not be mapped accurately. Also, the BC was distal to a telomere and its position in relation to the telomere could not be determined. Overall, therefore, these studies demonstrate that LouTat 1 and 1A utilize the same telomeric ES for transcription of their VSG genes.

Restriction map of LouTat 1 and 1A VSG ELC and BC gene chromosomal sites. The restriction maps of the LouTat 1 and 1A gene expression site and the 1A gene BC site are represented (restriction maps of LouTat 1, 1A, and 1.5 BC are the same). The cDNA sequence used as a VSG-specific probe is indicated by a short black bar. Restriction enzyme recognition sites are depicted, with telomere ends shown as vertical bars.
Discussion
The central focus of this study was upon the potential linkage of virulence to the telomeric ES for VSG genes in African trypanosomes. Specifically, we have asked whether two clonally derived, but biologically very different, trypanosomes express the same VSG gene or utilize the same VSG gene chromosomal ES. To address these questions we have cloned and characterized the cDNA sequences specific for VSG gene transcripts present in genetically related high- and low-virulence trypanosome populations and have also characterized the chromosome telomeric ES used for VSG gene expression in both.
The organisms T. brucei rhodesiense LouTat 1 and LouTat 1A express low and high virulence phenotypes, respectively. In the LouTat serodeme (VATs derived from LouTat 1), infection of animals with relatively low-virulence trypanosomes such as LouTat 1 gives rise to VATs (daughter cell populations displaying different VSG surface coat phenotypes) that express progressively higher levels of virulence as infection progresses (Inverso and Mansfield, 1983). This observation was exploited to derive a series of highly virulent subclones of LouTat 1 by rapid subpassage through immunocompromised mice; one of several subclones selected for further study was clone LouTat 1A, which caused death of mice in 3–4 days compared with 60–70 days for the parent clone from which it was derived (Inverso et al., 1988). Preliminary observations suggested that both LouTat 1 and 1A display similar surface coats, as seen from mAb- and pAb-binding tests and from partial protein sequence analysis (Inverso et al., 1988). In this study, nucleotide sequence analysis revealed that VSG gene cDNAs from LouTat 1 and 1A are precisely identical for both organisms and encode a molecule with conserved C terminal sequences common to type 1 VSG gene isotypes, as well as other conserved VSG-associated residues (Rice-Ficht et al., 1981; Reinitz et al., 1992; Blum et al., 1993). Variant specificity of the cDNA sequence was confirmed by northern blot hybridization to RNA from the homologous VATs LouTat 1 and 1A but not to RNA from the heterologous VAT LouTat 1.5 or to RNA from procyclic forms that do not express VSG genes. These results confirmed preliminary immunological and biochemical evidence that T. brucei rhodesiense clones LouTat 1 and 1A express the same VSG surface coat phenotype and VSG gene.
Genomic organization of the LouTat 1 VSG gene in the LouTat 1 serodeme was subsequently examined. In Southern blot analysis, VSG expressor populations LouTat 1 and 1A each contained two copies of the VSG gene, whereas nonexpressor populations contained a single copy of the gene. Because the restriction enzymes used did not cut within the VSG sequence, these restriction fragments represented discrete copies of the VSG gene. The additional band present in Southern blots of genomic DNA from expressor LouTat 1 and 1A populations but not heterologous VATs suggests that the VSG gene is transcribed in both populations following a gene duplication and transposition event that generates an ELC.
Telomere linkage of VSG gene ELCs has been well documented and it is known that VSG genes are expressed only from telomeric ES in bloodstream forms (Borst and Cross, 1982; de Lange and Borst, 1982; Myler et al., 1984a; Cross, 1990; Borst et al., 1998; Navarro and Cross, 1998; Navarro and Gull, 2001; Berriman et al., 2002; Hertz-Fowler et al., 2008). Additionally, basic copies of VSG genes are located at either internal or telomeric sites on chromosomes (Hoeijmakers et al., 1980; Pays et al., 1981a, 1981b, 1985; Majiwa et al., 1982; Williams et al., 1982; Bernards et al., 1984; Myler et al., 1984a, 1984b; Schopf and Mansfield, 1998). The single BC gene present in all heterologous VATs examined was shown to reside at a chromosome internal site location as shown by its relative insensitivity to Bal31 exonuclease digestion, despite the fact that some restriction fragments containing this gene copy were often of slightly different size. Variability in telomere size can be accounted for by the general growth of telomeric sequences at a rate of 6–10 bp per cell division and by occasional large deletions, which result in shortening of telomeres (Bernards et al., 1983; Van der Ploeg et al., 1984; Rudenko et al., 1996; Berriman et al., 2002; Sheader et al., 2003). The ELC of LouTat 1 and 1A VSG genes resides on an intermediate-sized chromosome, whereas the single BC present in all LouTat variants was contained on a larger chromosome. Restriction mapping of the LouTat 1 and 1A ES indicated that the VSG ELC expressed in both populations is located on the same telomeric ES, because restriction sites within the VSG gene, immediately 5′ to the VSG gene, and 5′ to the upstream barren region, are the same for LouTat 1 and 1A.
The significance of the present findings stems from our observation that T. brucei rhodesiense clones LouTat 1 and 1A differ dramatically in their virulence expression for mice and other species (Inverso et al., 1988) including nonhuman primates (Ndung'u and Mansfield, unpublished data). Because the VSG molecule plays a critical role in antigenic variation and because of the ability of the trypanosome to evade host immune responses during infection, we previously asked whether there was an association between expressed VSG molecules and virulence levels in the T. brucei rhodesiense LouTat 1 serodeme. Our preliminary observations, as well as unrelated studies by others (Musoke and Barbet, 1977; Mathias et al., 1990; Lucas et al., 1994; Magez et al., 1997, 1998), suggested that the VSG molecule could play a direct role in the relative expression of trypanosome virulence expression for a host. This suggestion was derived from our observation that all daughter cell trypanosome populations that expressed elevated virulence in an infected host were derived from a less-virulent trypanosome parent cell population that expressed a surface coat distinct from each of the virulent populations (Inverso and Mansfield, 1983). However, comparative examinations within the subsequently developed model system of T. brucei rhodesiense clones LouTat 1 and 1A that express the same VSG surface coat (based on earlier limited biochemical and immunological analyses and the results of this work) but differ dramatically in their virulence for mice, indicates that the VSG molecule itself is not associated with changes in relative virulence.
Also in this study, we have investigated further the potential relationship between VSG gene ES and virulence expression. As it is known that different ESAGs are coordinately transcribed with VSG genes as part of the polycistronic transcript generated within specific ES, and that ESAGs may regulate discrete aspects of trypanosome biology (Hoek and Cross, 2001; Berriman et al., 2002; Hoek et al., 2002), we searched for potential differences in the ES used by the VSG gene ELC in LouTat 1 compared with LouTat 1A. If the ES used by the VSG gene in two populations that differ in virulence was different, this finding would have provided a basis for further examination of the role of LouTat 1A ESAGs and their products in regulation of virulence. However, from the studies presented here it is apparent not only that the VSG molecules themselves are not virulence factors but also that the VSG gene and the VSG gene ES are not linked to virulence expression in the LouTat 1 serodeme of T. brucei rhodesiense. This conclusion is underscored by this work from our laboratory in which VATs derived from the highly virulent LouTat 1A expressed different VSG phenotypes and genes, as well as utilized different ES, but did not alter their virulence phenotypes (Inverso et al., 1988; Schopf and Mansfield, 1998; Mansfield and Paulnock, 2005; Mansfield, 2006). Overall, therefore, we predict that the virulence phenotype of African trypanosomes, as well as clonal changes in relative virulence for a host species, are regulated by genes mapping outside the VSG gene ES and are expressed independently of VSG gene expression.
Footnotes
Acknowledgments
The authors thank Hanna Filutowicz, John Barkei, Jim Schrader, and Karen Demick for their excellent technical assistance throughout this project. This study was supported by funds from the NIH (grants AI-22441, AI-39155, and AI-073346 to J.M.M., and AI-051421 and AI-048242 to D.M.P.).
Disclosure Statement
No competing financial interests exist.