
Abstract
Disruption of the X-chromosome fibroblast growth factor 16 (Fgf-16) gene, a member of the FGF-9 subfamily with FGF-20, was linked with an effect on cardiac development in two independent studies. However, poor trabeculation with lethality by embryonic day (E) 11.5 was associated with only one, involving maintenance in Black Swiss (Bsw) versus C57BL/6 mice. The aim of this study was to examine the potential influence of genetic background through breeding the null mutation onto an alternate (C57BL/6) background. After three generations, 25% of Fgf-16−/Y mice survived to adulthood, which could be reversed by reducing the contribution of the C57BL/6 genetic background by back crossing to another strain. There was no significant difference between FGF-9 and FGF-20 RNA levels in Fgf-16 null versus wild-type mice regardless of strain. However, FGF-8 RNA levels were reduced significantly in Bsw but not C57BL/6 mice. FGF-8 is linked to anterior heart development and like the FGF-9 subfamily is reportedly expressed at E10.5. Like FGF-16, neuregulin as well as signaling via ErbB2 and ErbB4 receptors have been linked to trabeculae formation and cardiac development around E10.5. Basal neuregulin, ErbB2, and ErbB4 as well as FGF-8, FGF-9, and FGF-16 RNA levels varied in Bsw versus C57BL/6 mice. These data are consistent with the ability of genetic background to modify the phenotype and affect embryonic survival in Fgf-16 null mice.
Introduction
The primary focus of this study was to explore the possibility that the genetic background can modify the embryonic lethal phenotype of Fgf-16 null mice. To this end, we have expanded the characterization of Bsw FGF-16 null mice to (1) investigate possible disruption during recombination of the immediately downstream and essential ATRX gene by assessing its expression, (2) examine the potential influence of genetic background on phenotype (including survival) through breeding the null mutation onto an alternate (C57BL/6) genetic background, and (3) compare RNA levels for members of the FGF-9 subfamily, which includes FGF-16, in different genetic backgrounds as potential targets for compensatory or modifying effects on the phenotype.
Materials and Methods
Mice
FGF-16 null mice were generated by homologous recombination in 129x1/SvJ ES cells as described previously (Lu et al., 2008a). The chimeric male founders carrying the targeted disruption in their germlines were backcrossed to either Bsw or C57BL/6 (C57) wild-type (WT) females. Subsequent generations were bred between heterozygous females (Fgf-16 +/−) and WT males from Bsw or C57 males. For the re-introduction of 129x1/SvJ genetic background, Fgf-16 +/− mice from the third generation of the C57 line were crossed with 129x1/SvJ males for another two generations. Mice were genotyped by polymerase chain reaction (PCR) with primers to detect the gene targeting construct and the presence of the Sry gene on the Y-chromosome as previously described (Lu et al., 2008a). All procedures were conducted in accordance with the guidelines of the Canadian Council on Animal Care and approved by the University of Manitoba Animal Protocol Review Committee, and conform with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Histology
Heart morphology was assayed as previously described (Lu et al., 2008a). Briefly, embryos were dissected from maternal tissue and fixed in zinc fixative, dehydrated, and embedded in paraffin. Paraffin sections were dewaxed, rehydrated, and stained with hematoxylin and eosin. Mutant embryos were somite-matched WT littermates.
RNA expression
Gestation was estimated based on the appearance of the vaginal copulation plug (representing E0.5). Total RNA was isolated from E10.5 fetal heart using the RNeasy Plus Mini kit (Qiagen, Mississauga, ON, Canada). Complementary DNA was reverse transcribed (RT) from 1 μg of total RNA using the QuantiTect Reverse Transcription Kit (Qiagen). Real-time quantitative RT-PCR (qRT-PCR) was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) in 25-μL reactions using the Power SYBR Green PCR Master Mix Kit (Applied Biosystems, Streetsville, ON) and specific primers for Fgf-8 (F, 5′-GCTCATTGTGGAGACCGATACTT-3′; R, 5′-TGGCAATTAGCTTCCCCTTCT-3′), Fgf-9 (F, 5′-ACTATCCAGGGAACCAGGAAAGA-3′; R, 5′-CTCGTTCATGCCGAGGTAGAGT-3′), Fgf-16 (F, 5′-GAGACAGTATTATGTGGCCCTGAA-3′; R, 5′-CTACTGGCCTTGGTAAAAAGTGAGT-3′), Fgf-20 (F, 5′-AGATGGTGCCAGGTCCAAAA-3′; R, 5′-ACATCAGTAGGTCTTTGTATAATTCTGGAA-3′) (Cohen et al., 2007), neuregulin-1β (NRG; F, 5′-AACAGCAGGCACAGCAGCCC-3′; R, 5′-AGGGGAGCTTGGCGTGTGGA-3′), epidermal growth factor receptor family member ErbB2 (F, 5′-GGTGGTGAGCTGACACTGG-3′; R, 5′-CACCATCAAACACATCGGAG-3′) (Kim et al., 2003), and ErbB4 (F, 5′-TGCGGGCAATATCTACATCA-3′; R, 5′-TTGAACACAGGTGGTTGCAT-3′) as well as GAPDH (F, 5′-TCACCACCATGGAGAAGGC-3′; R, 5′-GCTAAGCAGTTGGTGGTGCA-3′) RNAs. The baseline was set automatically, and the cycle threshold value was measured during the exponential phase of the amplification. Plasmids containing the amplicon sequences for GAPDH were used to quantitate DNA concentration and to determine the relative FGF RNA expression level in each sample (expression level = [ng FGF per μg cDNA]/[ng GAPDH per μg cDNA]). Tests were run in duplicate from at least three pairs of mice. Values (in arbitrary units) are expressed as mean ± standard error of the mean. Primers for the RT-PCR of ATRX are as follows: F, 5′-GGTTTTAGATGAAAATGAAGAG-3′; R, 5′-CACCATCTTCTTGCCATCTCTGTAG-3′. Statistical analysis was done by one-way analysis of variance with a post hoc (Tukey–Kramer multiple comparisons) test for multiple comparisons (significance, *p < 0.05), or by unpaired t-test for single comparisons (significance, † p < 0.05).
Immunohistochemistry
Day 11.5 embryos were dissected and fixed in 4% paraformaldehyde for 4 h on ice. Cryosections (7 μm) were pretreated with 3% H2O2 to deactivate endogenous peroxidase and incubated with Blocking Reagent (Roche, Laval, PQ, Canada) for 30 min at room temperature to reduce nonspecific binding of IgG. Avidin/Biotin blocking kit (Vector Laboratories, Burlingame, CA) was used subsequently to block endogenous biotin. Rabbit anti-mouse ATRX (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) was applied overnight at 4°C. The next day, biotin-conjugated goat anti-rabbit IgG (1:400 dilution; Jackson ImmunoResearch Laboratories, Westgrove, PA) was applied for 30 min, followed by incubation with VECTASTAIN ABC reagent (Vector Laboratories) for another 30 min. The signal was observed by 3,3′-diaminobenzidine substrate kit (Vector Laboratories) and counter stained with hematoxylin. The specificity of the antibody was confirmed by detection of the expected ATRX signal in an adult brain slice in cortex and hippocampus (Bérubé et al., 2005). Also, specificity of staining was checked routinely by omitting primary antibody and confirming a loss of signal.
Results
ATRX, the adjacent gene to Fgf-16 on the X-chromosome, is expressed in Fgf-16 null mice
ATRX is located adjacent to Fgf-16 on the X-chromosome in both mice and humans (Itoh and Ornitz, 2004; Nomura et al., 2006). Loss of ATRX leads to embryonic lethality during midgestation due to malformation of placenta (Garrick et al., 2006). To rule out any ATRX-related linkage effects that may lead to the embryonic lethality observed in our FGF-16 null mice with Bsw genetic background, we examined ATRX gene expression in both WT and null (Fgf-16 −/Y) embryos from Bsw backcrossings. RNA was isolated from the head (Hd) and heart (Ht) of null (−/Y) embryos and their WT (+/Y) littermates at E10.5. Samples were assessed by RT-PCR, with − RT controls, using specific primers to the ATRX and GAPDH RNAs in the same samples (Fig. 1A). Similar levels of ATRX RNA, relative to GAPDH RNA, were detected in Fgf-16−/Y versus WT embryos. ATRX protein expression was assessed in sections from null (−/Y) and WT (+/Y) (littermate) embryos at E11.5 by immunohistochemistry using specific ATRX antibodies and 3,3′-diaminobenzidine observation (Fig. 1B). ATRX was readily detected in comparable sections of both WT and null fetuses with similar expression patterns and levels (Fig. 1).
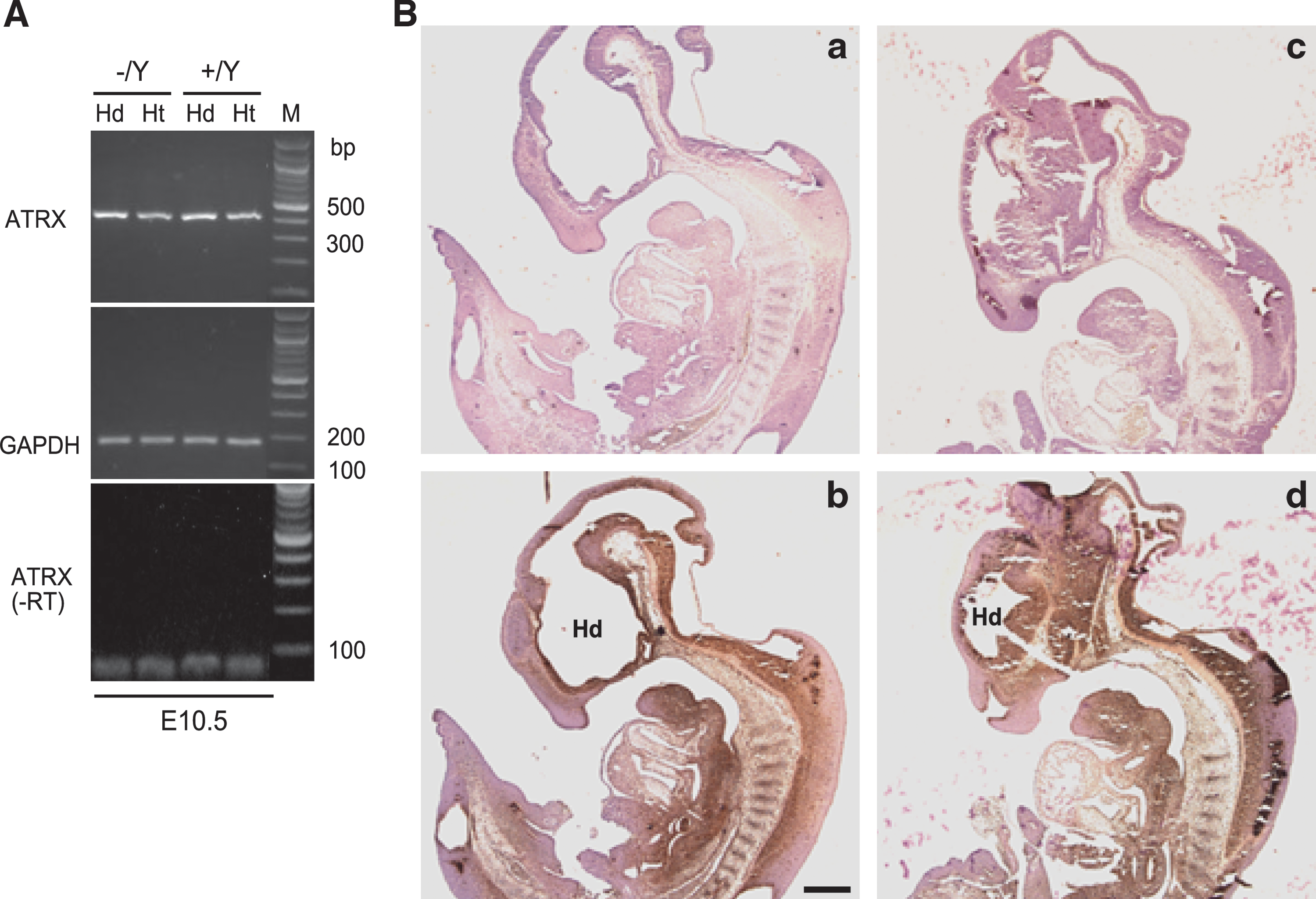
Expression of the ATRX gene in Fgf-16 null mice. (
Effects of genetic background on embryonic lethality and developmental defects in FGF-16 null mice
Fgf-16 null mice were generated through gene targeting in 129x1/SvJ ES cells (Lu et al., 2008a) and were subsequently backcrossed to either Bsw or C57BL/6 (C57) WT mice for at least six generations. The intent was to establish Fgf-16 mutant in both Bsw and C57BL/6 genetic background. In the Bsw backcrossing, all Fgf-16 null mice died in utero (Table 1). In contrast, with C57BL/6 backcrossing, after three generations approximately 25% of the Fgf-16−/Y mice survived to birth, based on theoretical genotype proportions (Table 2).
Breeding strategy: male founders that proved to transmit germline were bred with female wild-type Black Swiss mice to produce heterozygous (X+/X−) females. The heterozygotes were then backcrossed to the Black Swiss strain, and the offspring were genotyped (Lu et al., 2008a) at the time of weaning (3 weeks after birth). No differences were observed in the litter size between birth and weaning, indicating no genotype-related death after birth. The litter number and offspring number for each genotype at each generation are shown.
Breeding strategy: male founders that proved to transmit germline were bred with female wild-type C57BL/6 mice to produce heterozygous (X+/X−) females. Heterozygotes were then backcrossed to the C57BL/6 strain, and the offspring were genotyped (Lu et al., 2008a) at the time of weaning (3 weeks after birth). No differences were observed in the litter size between birth and weaning, indicating no genotype-related death after birth. The litter number and offspring number for each genotype at each generation are shown.
To validate the association of phenotypic shifting observed in the C57BL/6 backcrossed mouse line to genetic factors specific to the C57BL/6 strain, heterozygote female mice from the third generation of C57BL/6 backcrossing were mated with WT 129/SvJ mice. This was done to reduce the genetic contribution from C57BL/6 mice; 129/SvJ is the strain of mouse from which the ES cells originated. After two generations, the occurrence of the Fgf-16−/Y genotype in live births was no longer observed (Table 3). This phenotypic restoration of embryonic lethality further implicates the C57BL/6 genetic background in the survival of the FGF-16−/Y genotype to birth.
Fgf-16 heterozygotes at the third generation of C57BL/6 backcrossing were bred with either 129/SvJ(129) or C57BL/6(C57) mice for a further two generations, and the genotypes of the offspring of the second generation from the above breeding were analyzed at the time of weaning time (3 weeks after birth). No differences were observed in the litter size between birth and weaning, and thus no genotype-related death after birth. The litter number and offspring number for each genotype from the second generation of the above crosses are shown.
Characterization of FGF-16 expression during embryonic development, through knock-in of the β-galactosidase gene downstream of the Fgf-16 promoter region (Lu et al., 2008a) and detection of β-galactosidase activity, suggested expression of FGF-16 in the otic vesicle, pharyngeal arches, and head region at E9.5–E10.5 (Fig. 2). Endogenous FGF-16 expression is also found in heart at this stage by tissue dissection and RT-PCR (Lu et al., 2008a). Thus, this evidence of Fgf-16 promoter activity in regions of the head and heart links FGF-16 expression to both craniofacial and cardiac development. This is consistent with the pattern of endogenous FGF-16 RNA detected in the developing ear, pharyngeal arch, and heart by others (Wright et al., 2003; Lavine et al., 2005; Chapman et al., 2006; Hatch et al., 2009). The morphology of Fgf-16−/Y embryos and somite-matched WT littermates was analyzed at various developmental stages from F4 to F6 mice in both Bsw and C57BL/6 backgrounds. At E9.5, mutants showed indistinguishable heart morphology from the WT in both genetic backgrounds, whereas by E11.5 there was visible dilation of the common (or primitive) ventricle with compromised trabeculae in BswFgf-16−/Y embryos, and gross examination of embryos revealed consistent craniofacial malformation (Lu et al., 2008a). Similar morphological changes to the head (Fig. 3f) and heart (Fig. 4c) regions were also observed among the C57Fgf-16−/Y embryos. However, in contrast to BswFGF-16−/Y , a range of phenotypes was identified among C57Fgf-16−/Y embryos. In light of the consistent defects observed by E11.5 in Bsw Fgf-16 null mice, this time point was used not only to screen for possible changes between strains but also to provide representative images of the range of phenotypes observed. These included examples with comparatively normal versus abnormal craniofacial (Fig. 3d vs. e and f ) or heart (Fig. 4e vs. b, c, and f ) development. For those C57Fgf-16−/Y mice that survived and were assessed after birth, there was no obvious defects compared to their WT littermates, including in the mouth and jaw, and it was noted that they feed, grow, and breed normally.
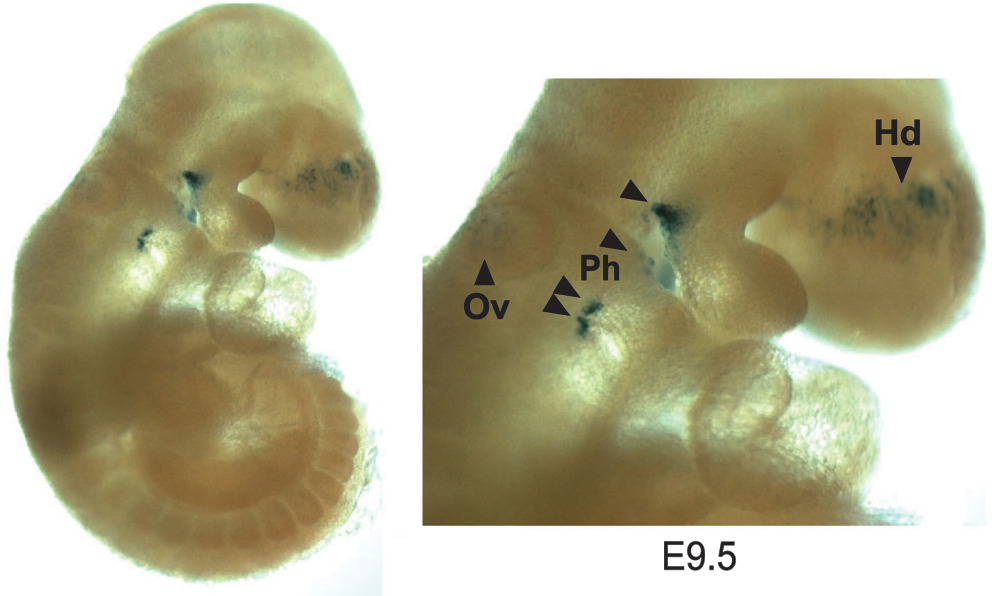
Pattern of FGF-16 expression as revealed through detection of β-galactosidase activity following introduction of the β-galactosidase gene downstream of the Fgf-16 promoter through recombination in a BswFgf-16−/Y mouse at E9.5. Arrowheads indicate staining of the otic vesicle (Ov, ear), pharyngeal arch (Ph; derived structures include upper/lower jaw, cardiac outflow tract, and heart, thymus, and parathyroid), and face (Hd, vicinity of optic eminence). Bsw, Black Swiss.
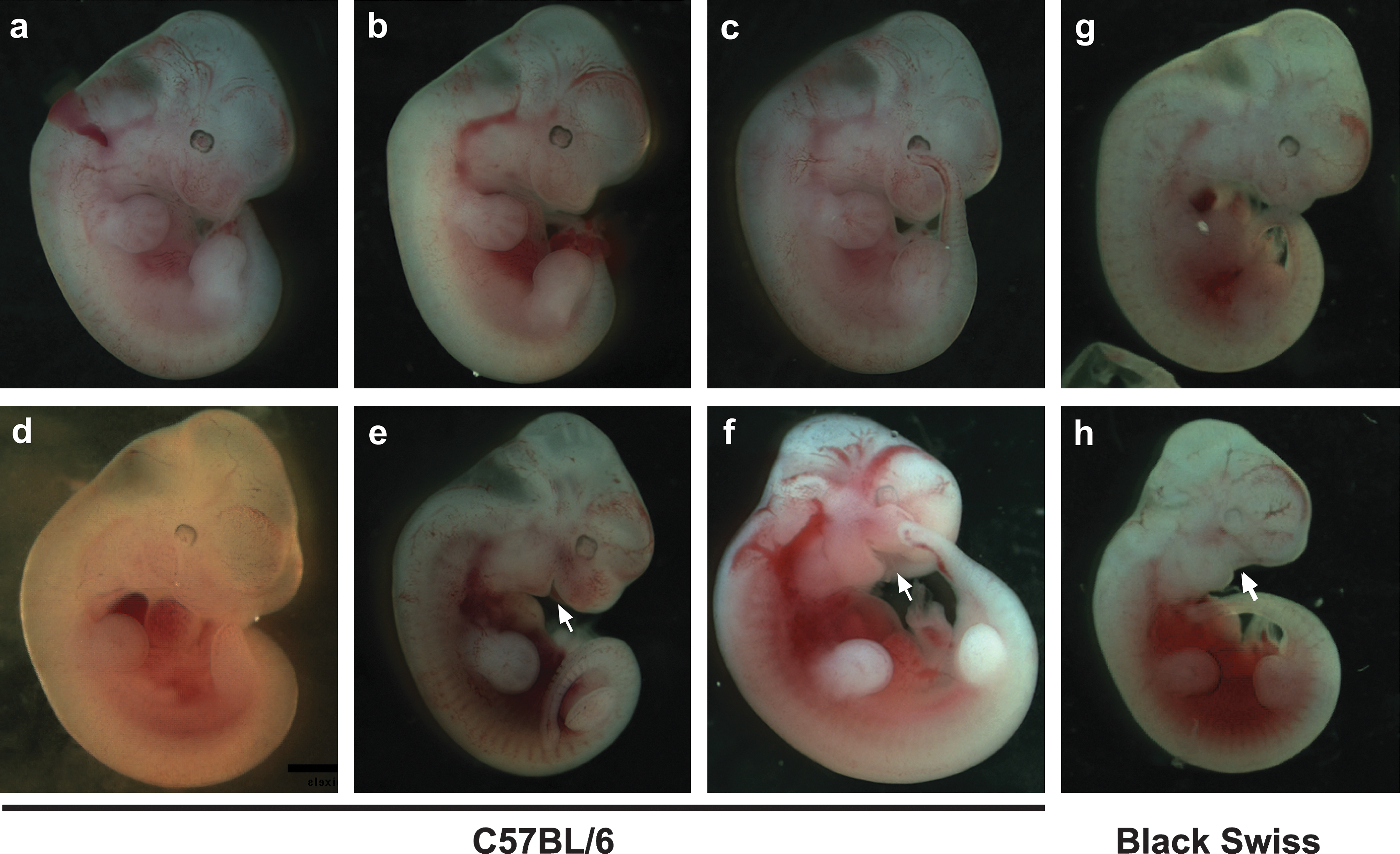
Whole-mount preparations of WT (
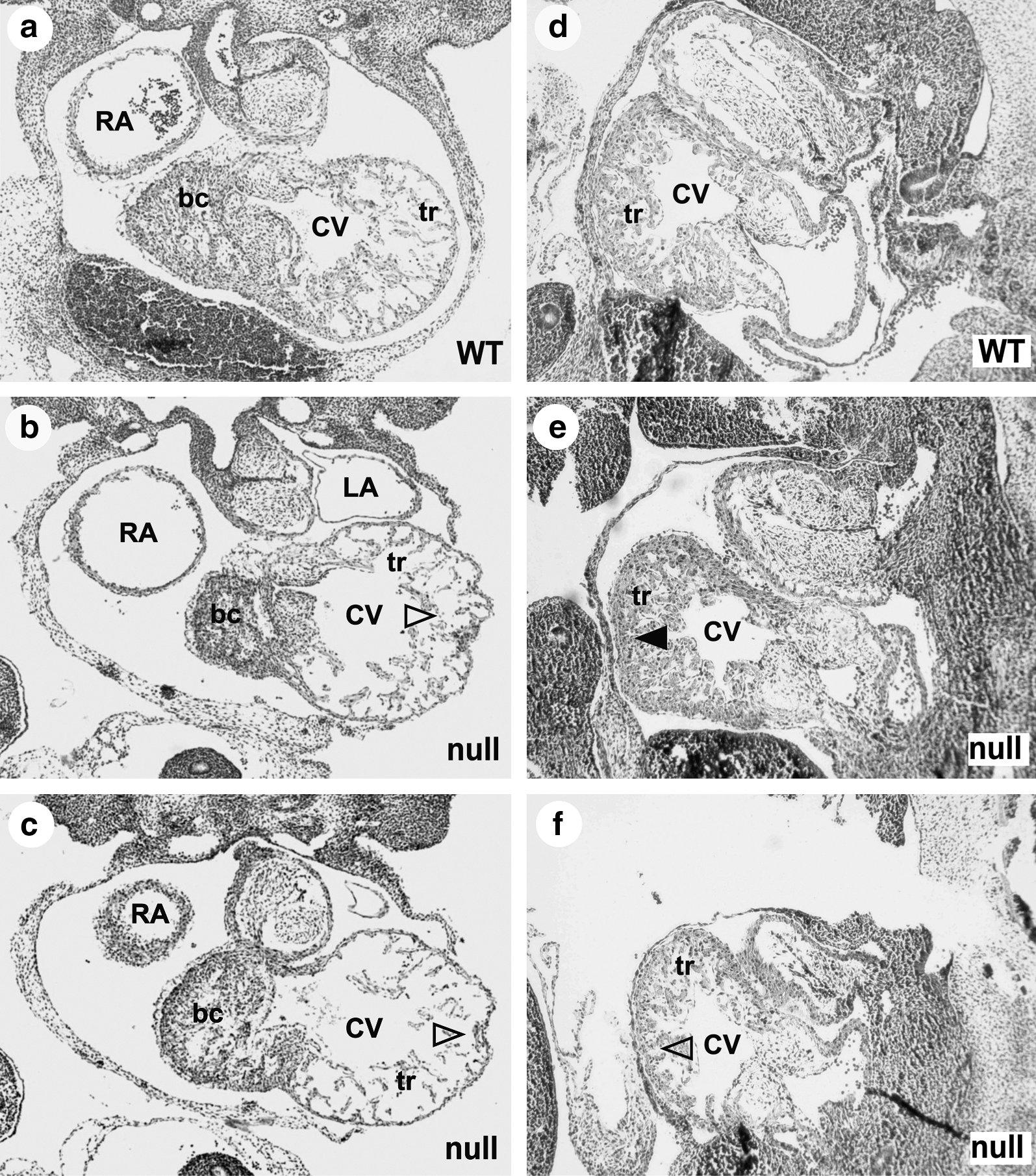
Histological assessment of the heart phenotype in C57BL/6 Fgf-16 null mice at E11.5. Transverse (
A comparison of FGF-9 subfamily and NRG/epidermal growth factor receptor family member (ErbB2 and ErbB4) RNA levels at E10.5 in Bsw and C57BL/6 mice
It is possible that other members of the FGF family, and specifically the FGF-9 subfamily, might compensate for the lack of FGF-16. FGF-9 and FGF-20 share >70% amino acid sequence similarity with FGF-16 in some regions compared to <50% when compared to other FGF family members (Kirikoshi et al., 2000) as well as some distinct but also overlapping patterns of expression during embryonic heart development (Lavine et al., 2005). As such there is a greater potential for FGF-9 and/or FGF-20 (versus other FGF family members) to compensate for a Fgf-16 null mutation (potential issue of redundancy). Levels of RNA for each member of the FGF-9 subfamily (FGF-9, FGF-16, and FGF-20) were assessed at E10.5 in BswFgf-16−/Y and C57Fgf-16−/Y mice and their WT littermates by real-time RT-PCR. In addition, FGF-8 RNA levels were tested since it has been linked to anterior heart development and is also expressed at E10.5 in the heart (Waldo et al., 2001). Relative levels of FGF-8, FGF-9, and FGF-16, but not FGF-20, RNA were significantly different between these two strains in WT mice (Fig. 5); FGF-8 RNA levels appeared lower in C57BL/6 mice, whereas FGF-9 and FGF-16 were higher. However, relative FGF-9 RNA levels were 50–250 times greater than FGF-16 and/or FGF-20 levels in C57BL/6 compared to Bsw. In terms of the effect of Fgf-16 gene disruption, there was no significant effect on FGF-9 or FGF-20 RNA levels in either mouse strain. However, FGF-8 RNA levels were reduced (∼2-fold) significantly in Bsw, but not in C57BL/6, null mice.
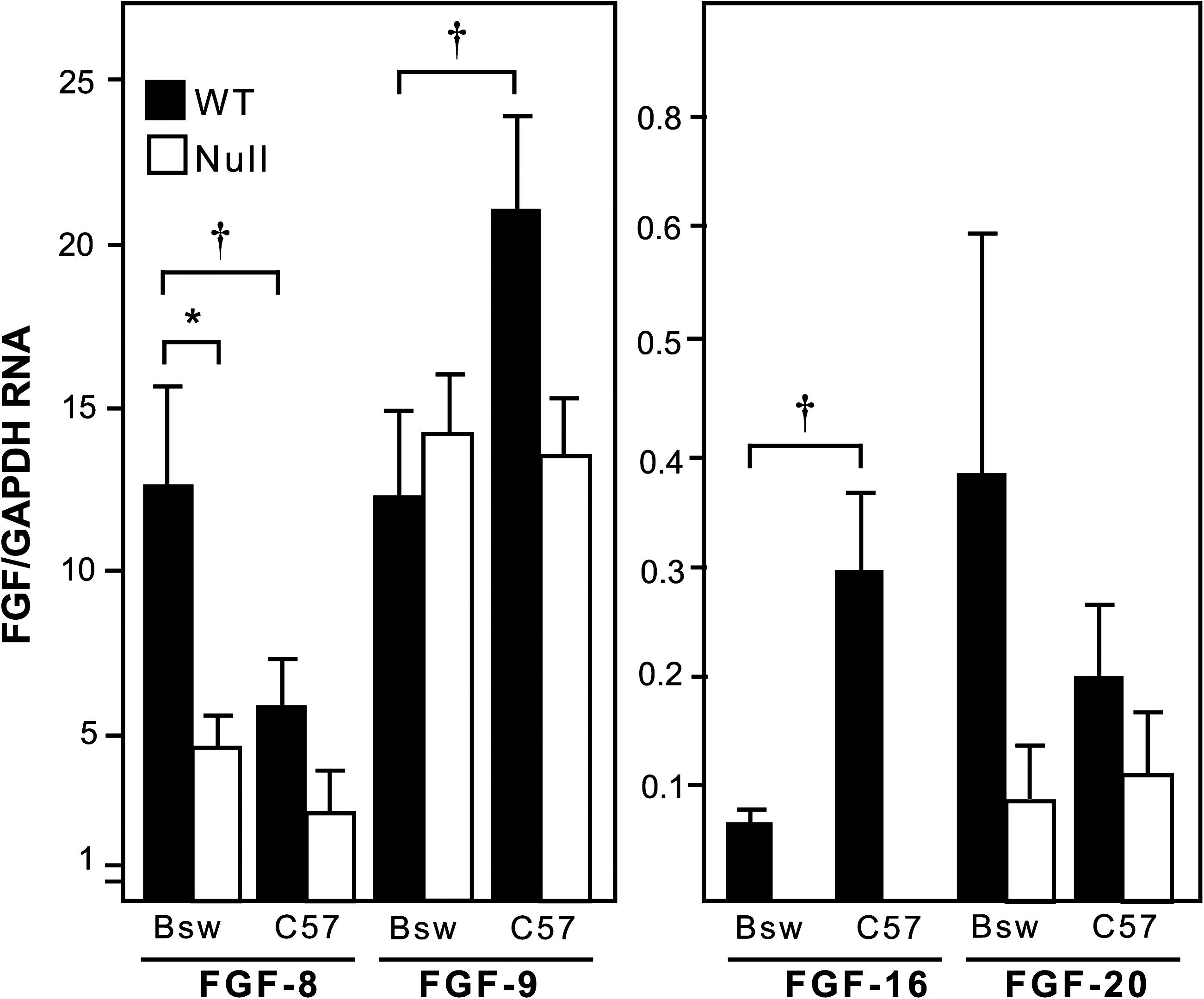
Relative levels of FGF-8, FGF-9, FGF-16, and FGF-20 RNA in the heart of different WT and Fgf-16 null mice strains at E10.5. Heart RNA was isolated and assessed by real-time RT-PCR using specific primers to Fgf-8, Fgf-9, Fgf-16, and Fgf-20 as well as GAPDH, which was used to normalize the data. All results are expressed as means (in arbitrary units), and error bars indicate standard error of the mean (*p < 0.05, n = 3). Different scales are used for FGF-8 and FGF-9 versus FGF-16 and FGF-20 because of the difference in relative expression. Comparisons of FGF RNA levels within a mouse strain were assessed by analysis of variance with post Tukey–Kramer multiple comparisons test (*p < 0.05), and individual FGFs compared in the two strains were assessed by unpaired two-tailed t-test († p < 0.05).
NRG/ErbB signaling plays an important role in murine cardiac development, and mutations of the ErbB2 and ErbB4 receptors in particular have been associated with defective trabeculae formation and embryonic lethality around E10.5 (Kim et al., 2003; Iwamoto and Mekada, 2006; Pentassuglia and Sawyer, 2009). Poor trabeculation was also observed in hearts of BswFgf-16−/Y mice at E10.5 (Lu et al., 2008a). Thus, levels of RNA for NRG-1β as well as ErbB2 and ErbB4 receptors were assessed at E10.5 in BswFgf-16−/Y and C57Fgf-16−/Y mice and their WT littermates by real-time RT-PCR, as possible targets of FGF-16 during development. Relative levels of NRG, ErbB2, and ErbB4 RNA were all significantly higher in WT C57BL/6 versus Bsw mice (Fig. 6). However, there was no significant effect of the Fgf-16 null mutation on relative NRG, ErbB2, or ErbB4 RNA levels in either mouse strain.

Relative levels of NRG-1 and epidermal growth factor receptor ErbB2 and ErbB4 RNA in the heart of different WT and FGF-16 null mice strains at E10.5. Heart RNA was isolated and assessed by real-time RT-PCR using specific primers to NRG, ErbB2, and ErbB4 as well as GAPDH, which was used to normalize the data. All results are expressed as means in arbitrary units as indicated (except for ErbB4/Bsw and ErbB4/C57 that are ×10−5 and ×10−3, respectively) and error bars indicate standard error of the mean (n = 3–7). Comparisons of RNA levels within a mouse strain were assessed by analysis of variance with post Tukey–Kramer multiple comparisons test, and individual RNA compared in the two strains was assessed by unpaired two-tailed t-test († p < 0.05 and ††† p < 0.005). NRG, neuregulin.
Discussion
In this study, we show for the first time that genetic background can influence the embryonic lethal phenotype resulting from a Fgf-16 null mutation. Consistent and severe craniofacial and heart defects, as well as associated embryonic lethality, are observed in mice that are backcrossed to the Bsw line (BswFGF-16−/Y ) (Lu et al., 2008a). In contrast, while a similar phenotype was observed initially in a mixed genetic background (C57BL/6 and 129/SvJ), these defects were rescued in a percentage of the mice after three generations of backcrossing to C57BL/6 mice. The surviving (Fgf-16) null mice resulting from C57BL/6 backcrossing grow to adulthood, breed normally, and display no obvious morphological defects. This includes regions of the head that displayed evidence of Fgf-16 promoter activity such as the developing ears and jaw; a role for FGF-16 in the developing ear has been indicated in previous chicken and mouse models (Chapman et al., 2006; Hatch et al., 2009). Our results following C57BL/6 backcrossing are consistent with those of others who report apparently normal and fertile Fgf-16 null mice in a C57BL/6 genetic background (Hotta et al., 2008). In addition, we have shown that embryonic lethality in our BswFgf-16−/Y offspring can be restored by reducing C57BL/6 genetic background via the re-introduction of the 129/SvJ strain. This further implicates strain-specific genetic components from C57BL/6 mice in the survival of Fgf-16 null mice. In addition, there was no evidence that ATRX gene expression (the most closely linked gene to Fgf-16 on the X-chromosome) was affected in BswFgf-16−/Y mice, making it unlikely that the phenotype observed in the present study comes from disruption of ATRX expression or more global changes to the X-chromosome during homologous recombination. Our data instead support the hypothesis that the genetic network (and/or its regulation) is somehow variable in mice with different genetic backgrounds, resulting in different strains displaying different capacities to functionally substitute for the loss of FGF-16.
Genetic background can affect the phenotype of a given mutation, and has been demonstrated by both targeted disruption (Strunk et al., 2004; Astrof et al., 2007) and overexpression of a transgene (Krezowski et al., 2004; Sebastiani et al., 2006). This has focused attention on the effects of modulating genetic background on Mendelian traits, particularly in regard to interpreting results, as more and more genetically modified mice are developed to assess gene function. The phenotype found in any knock-out mouse is the result of genes present in the background strain working in combination with the effect of the loss of the gene of interest. The effect of genetic background on the Fgf-16 null mutation is interesting by virtue of the magnitude and completeness of the rescue. The observations in our present study and the data from another group (Hotta et al., 2008) indicate that near-normal development of cardiac and craniofacial structures can occur in a C57BL/6 genetic background in spite of Fgf-16 gene disruption. This suggests that none of the cells or structures derived from the heart and pharyngeal arches depend exclusively on FGF-16 for their expansion and/or differentiation. In contrast, the effects of genetic background and the presumed effect of modifier gene(s) in this context appear to be significant. FGF signaling regulates multiple facets of embryonic morphogenesis, including cell proliferation, differentiation, and migration. However, most Fgf null mice are viable with subtle phenotypes (Itoh, 2007), suggesting that a redundant effect is possible in the FGF family.
Genetic factors that modulate the effect of the gene of interest, the modifier gene, may function through one of numerous mechanisms, including suppression or enhancement of gene expression involved in physiological or pathological pathways (Linder, 2006). Candidates for a compensatory response to an FGF-16 deficiency include other members of the FGF-9 subfamily. Each of the three members of the subfamily was found in midgestation embryonic heart at E10.5 and was able to induce DNA synthesis in cardiac myocytes (Lavine et al., 2005). Thus, a temporal and/or spatial change in the pattern of FGF-9 and/or FGF-20 expression in response to the lack of FGF-16 is feasible to explain the rescue phenotype. In support, relative FGF-9 RNA levels were significantly higher in the C57BL/6 strain than in Bsw. Although we did not detect a significant difference between Bsw and C57BL/6 FGF-9 or FGF-20 RNA levels in our null animals, levels were only assessed at one time point, E10.5 (and may have missed a key stage of development due to changes in the timing of events in the absence of FGF-16). Interestingly, FGF-8 RNA levels were reduced significantly in Bsw null mice, but not in those backcrossed to C57BL/6 mice. FGF-8 is involved in anterior heart field development, and a deficiency leads to craniofacial and heart defects. The relative basal level of FGF-8 RNA was also found to be lower in C57BL/6 versus Bsw mice, and an inverse relationship with FGF-16 RNA levels is suggested, which raises the possibility that Bsw mice are more sensitive to a decrease in FGF-8 levels. However, the level of FGF-8 RNA at E10.5 in 129/SvJ mice was not different from C57BL/6 (data not shown), although backcrossing to 129/SvJ mice did result in embryonic lethality, suggesting a different temporal/special combination of factors might exist in 129/SvJ for normal embryonic development.
There was no significant effect of the Fgf-16 null mutation on NRG, ErbB2, or ErbB4 RNA levels at E10.5. The link between NRG/ErbB signaling in cardiac development has been well established through the use of null mutation and transgenic mouse studies (reviewed in Falls, 2003; Iwamoto and Mekada, 2006; and Pentassuglia and Sawyer, 2009). This was particularly intriguing in relation to the poor trabeculation observed in BswFgf-16 null mice at E10.5, which is more pronounced at E11.5 (Lu et al., 2008a). Lack of NRG is known to limit cardiac cushion and trabeculae development between E9.5 and E10.5, and null mutations of either NRG receptor ErbB2 or ErbB4 disrupt trabeculation. Thus, based on the timing of the effects on trabeculation, an effect of NRG/ErbB levels/signaling on FGF-16 levels/signaling might be predicted rather than the other way round, and if so, the lack of effect of the Fgf-16 null mutation on NRG/ErbB RNA levels in both mouse strains would not be unexpected. Nonetheless, the possibility that higher levels of the NRG/ErbB signaling pathway in the C57BL/6 line contributes to the surviving phenotype in Fgf-16 null mice cannot be ruled out. Further, the potential for crosstalk between FGF-16 and NRG/ErbB signaling is intriguing not only in terms of embryonic development but also in the context of the adult heart, where Erb2 and Erb4 (as well as Erb1) like FGF-16 are expressed in the myocardium and have been linked to cardiac stress (Zhao et al., 1998; Pentassuglia and Sawyer, 2009; Sofronescu et al., 2010).
Breeding into a C57BL/6 background with relatively few (four to eight) back crossings has been linked to phenotypic changes as well as rescue of embryonic lethality in other genetically disrupted mouse models (Hao et al., 2005; Lygate et al., 2009). The observation of a range of (facial and heart) phenotypes in the progeny backcrossed to C57BL/6 mice reported here, as well as the observations made with regard to FGF-8 RNA levels, suggests that there are modifications to expression of multiple genes, not a single gene, likely due to heterogeneity of modifiers. This was also the conclusion reached following an unsuccessful attempt to localize modifiers in the case of ErbB signaling through whole-genome scans (Strunk et al., 2004); the Fgf-16 null mutation, like disruption of ErbB2 and ErbB4 signaling, is associated with negative effects on cardiac development and trabeculation. However, in a subsequent study the effect of intercrossings on lethality associated with disruption of ErbB signaling was done with multiple mouse lines, including C57BL/6 mice as well as Swiss-related inbred strains (Strunk et al., 2004). The major effect of modifying alleles in crossings with C57BL/6 mice was seen associated with events at E10.5–E13.5, in contrast to Swiss-related strains where the major effect on phenotype was seen later in gestation (E14.5–E18.5). This concept is consistent with our studies, which demonstrate the apparent rescue of a potentially lethal Fgf-16 null mutation by E11.5 in an increased C57BL/6 genetic background. Thus, the evidence is consistent with a network responsible for establishing sufficient compensation in the absence of FGF-16 that is modulated by genetic background, thus leading to different resiliency.
Footnotes
Acknowledgments
The authors acknowledge Ms. Karen Detillieux's contribution to the development of this article, and thank Ms. Agnes Fresnoza for technical assistance. This work was supported by a grant from the Canadian Institutes of Health Research (MOP-62742). S.Y.L. was the recipient of a fellowship from the Manitoba Institute of Child Health.
Disclosure Statement
No competing financial interests exist.