
Abstract
The myostatin (MSTN) gene region encompassing the 5′UTR and part of intron I was sequenced in animals of two herds of Latvian Darkhead sheep to extend data on the ovine MSTN gene polymorphism and to provide information useful for local breed conservation. Two and four polymorphic loci were revealed in the 5′UTR and intron I. Four and five local haplotypes were constructed, respectively. The genotyping data obtained and that previously reported for the same genomic region were combined in one dataset for the haplotype analysis. Recombination events were detected between loci (c.−40, c.−37) in the 5′UTR and (c.373+18, c.373+101) and (c.373+101, c.373+241) in intron I. Single-nucleotide polymorphisms at c.373+249 and c.373+323 appear to be involved in the strong linkage (p < 0.01). Linkage blocks (c.373+241, c.373+243) and (c.373+241, c.373+259) were revealed at nominal (p < 0.05) level of probability. Haplotype-specific patterns of the transcription factor binding sites predicted in silico were constructed to evaluate a putative functional significance of the particular alleles and haplotypes. A nucleotide at c.373+18 was shown to influence the pre-mRNA secondary structure. DNA curvature predicted in silico for allele c.373+101C was proven experimentally. A possible impact of the particular polymorphisms on the transcription and/or splicing efficiency is discussed.
Introduction
Haplotypes could be more informative in the genetic diversity and association analysis, as linkage disequilibrium between polymorphic loci can be taken into account. Three two-locus haplotypes were identified in the 473-bp gene region encompassing exon I and intron I in NZ Romney (Zhou et al., 2008). Four haplotypes encompassing three polymorphic loci in the 5′UTR and gene coding region were analyzed in BT (Kijas et al., 2007). Hickford et al. (2010) described five haplotypes encompassing nine polymorphic loci in intron I. An association was found between the most frequent haplotype (allele A) and decreased leg, loin, and total yield of lean meat in NZ Romney.
Little is known about the functionality of the structural variations in the MSTN gene. From those located in the gene noncoding regions, only SNP c.*1232G>A was shown to create a target site for microRNAs mir1 and mir206 highly expressed in skeletal muscles. Binding of the microRNAs could cause translational inhibition of the MSTN gene and, consequently, contribute to muscular hypertrophy in Texel sheep (Clop et al., 2006). Several motifs potentially important for the transcriptional regulation were also identified in the promoter of the ovine MSTN gene (Du et al., 2007).
The above data suggest that a larger variety of breeds should be studied to fully characterize the spectrum of variations in the ovine MSTN gene. The functionality of the particular variations and haplotype configurations should be included in the analysis to better understand the mechanisms of the mutational effects on the phenotype.
The aims of the present study were (1) to extend currently limited data on sheep MSTN gene polymorphism, (2) to analyze single locus and haplotype diversity in the Latvian Darkhead (LD) breed to provide useful information for breed conservation, and finally, (3) to evaluate the possible functional significance of the MSTN gene polymorphism.
Materials and Methods
Breed description: sampling and DNA extraction
The LD breed originated from the cross performed in the beginning of the 20th century between local Latvian coarse wool, Shropshire, and Oxford Down breeds and was registered as a Latvian local breed in 1937. Crosses with animals of Finnish Landrace, Ile-de-France, and Texel breeds were used, respectively, in the seventies and eighties of the last century to improve breed fertility and meat quality. Emphasis on both meat and wool quality selection resulted in a number of LD breed features (
Reference alleles (first indicated nucleotide) are given according to the sheep MSTN gene sequence information (GenBank accession numbers: AY918121, AF393618, and AM992883).
LLU, Latvia University of Agriculture; PR, private.
DNA was isolated from 500 μL of blood of each animal by using a K0512 Genomic DNA Purification Kit (Fermentas). The research was approved by the LLU review board.
Sequence information
Sequence information for sheep MSTN gene (AY918121, AM992883, and AF393618) available in GenBank was used for genomic region reconstruction and primer design. MSTN gene sequence information for Capra hircus (EF591039), Bos taurus allelic variants (AB076403, AF320998, and AY850105), Equus caballus (NC_009161), Sus scrofa (EF490990), and Homo sapiens (NG_009800) was used in the comparison of sequences. Numbering of the nucleotides was conducted according to the nomenclature system (
DNA amplification and sequencing
Two primer pairs (Fig. 1 and Table 2) were used to amplify gene fragments of 809 bp and 645 bp (Fig. 1). Amplicons were sequenced in both forward (primers Ex1sF and AMOsF) and reverse (primers Ex1sR and AMOsR) directions (Table 2 and Fig. 1). Amplification and sequencing were repeated two or three times per amplicon/animal to obtain clearly readable sequences overlapping each other in 153 bp (Fig. 1) and provide sequence information on the 1037-bp MSTN gene region encompassing part of the promoter, the 5′UTR, exon I, and part of intron I. The microsatellite (MS)-containing fragment of the 270 bp was synthesized using MSF and AMXaR (Table 2 and Fig. 1) primers. Primer design was performed using the Primer3 web tool (
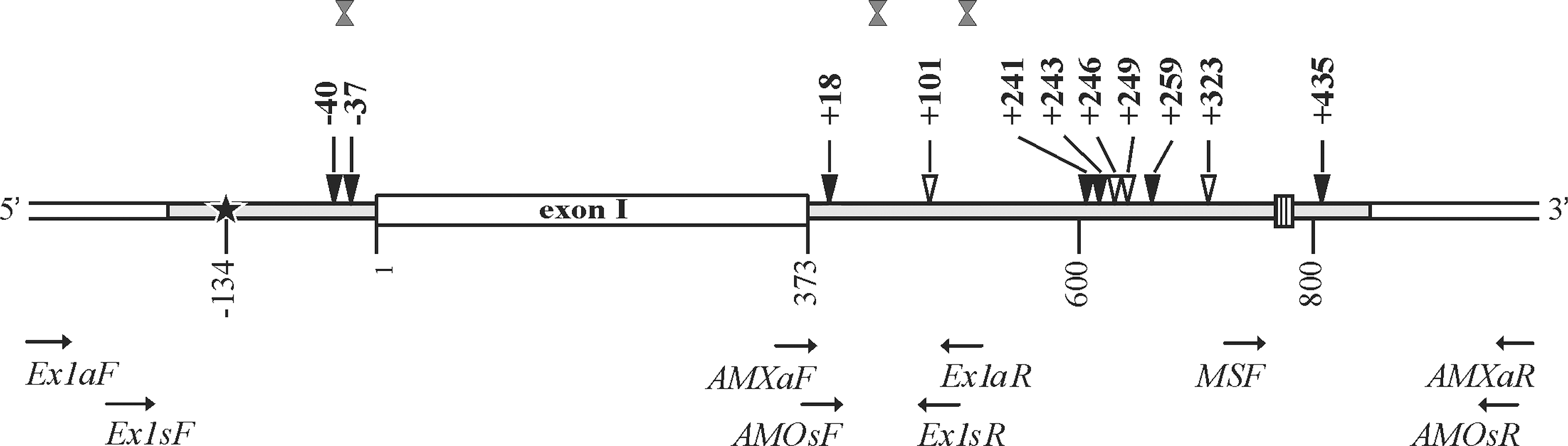
Scheme of the analyzed region of the myostatin (MSTN) gene and the strategy of the experiment. Sequenced 5′UTR and intron I gene regions are marked in gray. Horizontal arrows indicate the positions of primers used in amplification and sequencing. 5′UTR start and microsatellite locus are marked by black star and shaded box, respectively. Loci polymorphic in Latvian Darkhead (LD) and monomorphic in LD but polymorphic in other breeds (Clop et al., 2006; Hickford et al., 2010) are indicated by black and white triangles, respectively. Recombination sites are indicated by double gray triangles.
In the primer name, letters “a” and “s” indicate the primer sequences used for amplification and sequencing, respectively.
Electrophoretic mobility assay
Bent DNA analysis was performed using the previously described protocol (Pasero et al., 1993). Two intron I fragments including the 645 and 270 bp amplicons were analyzed in two electrophoretic systems: 1% conventional agarose gel and 12% polyacrylamide (PAAG; at 0.5 V/cm and 4°C). Gels were stained with ethidium bromide.
The mobility pattern of each fragment was obtained by calculating the R-value, that is, the ratio of apparent to real fragment size observed in each gel system. Values 1.09 ≤ R ≤ 0.90 were considered to indicate alteration of mobility (Gimenes et al., 2009).
Data management and analysis
Sequences obtained in this study were deposited in GenBank under accession numbers FJ809775 to FJ809783. ClustalW (
DnaSP4.5 software (
Results
Polymorphism discovery and genetic diversity
Two and four loci in the 5′UTR and intron I (Table 1 and Fig. 1), respectively, were found to be parsimony variable sites with two variants. The locus at c.373+435 was presented by nucleotide A in all animals analyzed as well as a previously unidentified nucleotide at c.373+434. The presence of the homozygote (Gen 1, Gen 2) and single-locus heterozygote (Gen 3, Gen 4) genotypes suggests three 5′UTR (C−40T−37, A−40T−37, and C−40C−37) and two intronic (Hap 1 and Hap 2) haplotypes. Multiloci heterozygote genotypes suggest additionally the A−40C−37 5′UTR haplotype and intronic Hap 3, Hap 4, and Hap 5 (Table 3). The Hap 1, Hap 2, and Hap 5 correspond to haplotypes (alleles) A, B, and E, respectively, described by Hickford et al. (2010), with only one exception, namely SNP at the c.373+101 (Table 3). SNPs and haplotype sequences observed in LD were submitted to GenBank under accession numbers rs119102823–rs119102830 and FJ809775–FJ809783, respectively.
Symbols (*), (#), and (+) mark the pairs of haplotypes identical at all loci except c.373+101. Symbol (^) indicates the nucleotides observed in Texel with frequency 0.99 (Clop et al., 2006).
LD, Latvian Darkhead; Tex, Texel; ATex, Australian Texel; NZ, New Zealand Romney; DD, Dorset Down; M&P, Merino&Polwarth; SD, Southdown.
The genotyping data obtained in our study and that previously reported (Clop et al., 2006; Hickford et al., 2010) were combined in one dataset in the DnaSP analysis. The strongest linkage (p < 0.01) was revealed between the c.373+249 and c.373+323 loci. Two other blocks (c.373+241, c.373+243) and (c.373+243, c.373+259) were predicted with a nominal (p < 0.05) level of probability. One recombination event was detected in the 5′UTR (Fig. 1).
Gen 1 (five animals from eight; Table 1), the C−40T−37 5′UTR haplotype, and intronic Hap 1 appear to be predominant in the LLU herd (frequency: 0.84 and 0.81, respectively) in contrast to the PR herd, which was found to be more genetically heterogeneous (Table 1). Predominant in LD, the C−40T−37 haplotype of the 5′UTR was previously observed as most frequent in BT (Clop et al., 2006) (Table 3). The configuration A−40T−37 found in LD exists also in the goat, cattle, horse, and swine genes (Fig. 2A). The A−40C−37 haplotype seems to be a specific feature of the human gene (Fig. 2A). The high sequence similarity in the 5′UTR of sheep, goat, and cattle genes (Fig. 2A) calls forth the common cluster in the phylogenetic tree (Fig. 3A).

MSTN gene partial sequence alignment between species. (
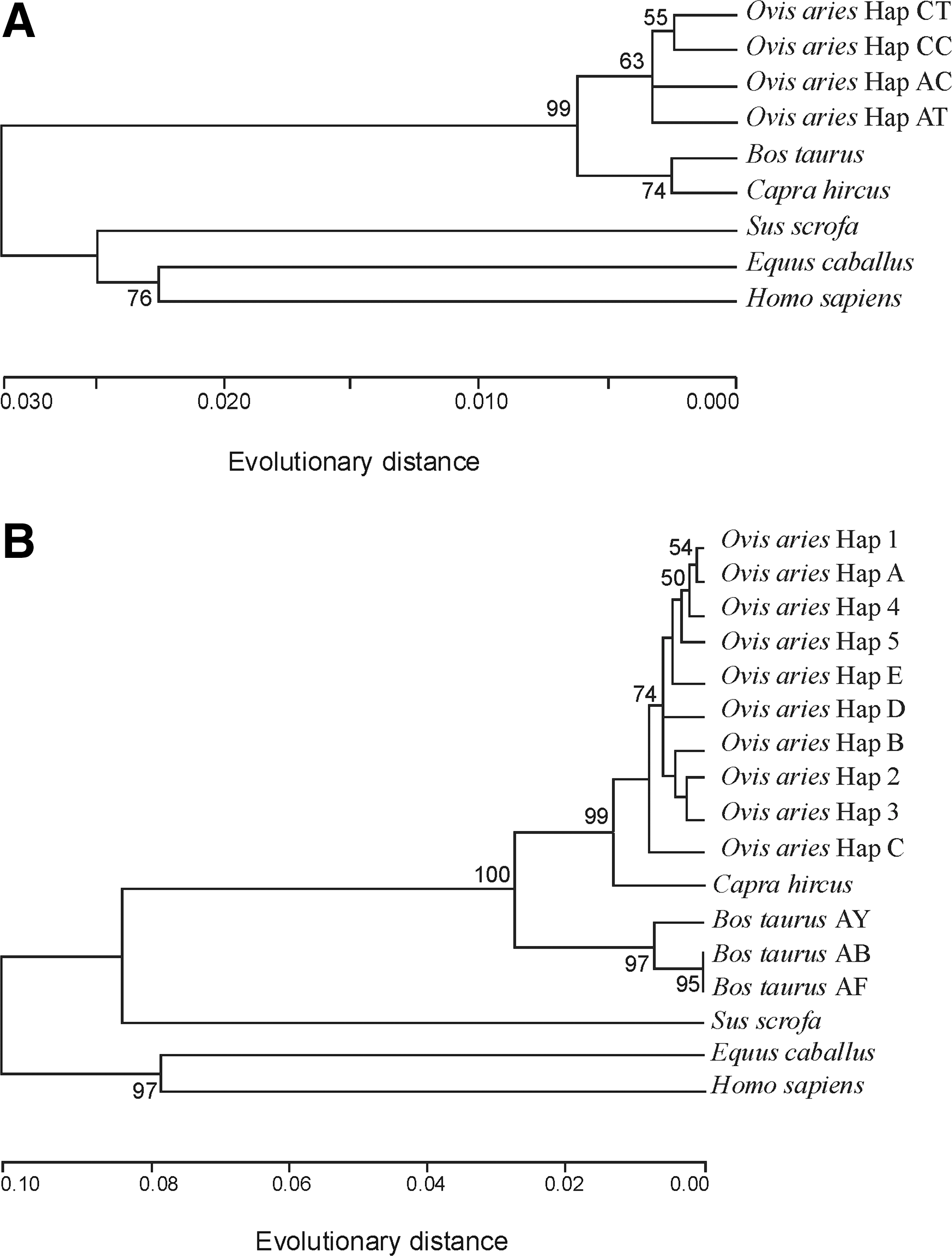
Phylogenetic trees of the MSTN gene noncoding regions. (
Taking into account our data and that reported by Hickford et al. (2010), at least 10 haplotypes of intron I could be now distinguished in sheep (Table 3). Hap 1 and Hap A are obviously the most frequent haplotypes in LD and NZ Roman breeds, respectively. Compared with the 5′UTR, the intronic sequences have less interspecies similarity. The loci found to be polymorphic in sheep appear to be monomorphic in the goat, cow, horse, swine, and human genes, and the same nucleotides are often found in these positions in the latter species (Fig. 2B–F). In the phylogenetic tree, the evolutionary distances within and between species are larger compared with the 5′UTR (Fig. 3). Goat unites the two sheep subclusters defined by the nucleotide at c.373+243 (Fig. 3B). Cattle form their own subcluster. Difference between the two phylogenetic trees suggests difference between the 5′UTR and intron I in evolutionary and breeding history.
Functional significance of MSTN gene variation
Figure 4A and B illustrates the haplotype-specific patterns of the TFBSs coupled with nucleotide variations within the 5′UTR and intron I. A detailed TF family/matrix description is given in Table 4. Allele A−40 defines the 5′UTR sequence similarity to binding sites with zinc finger protein ZNF35 (AC and AT haplotypes). Nucleotide C−37 creates the binding site to thyroid TTF1 factor (AC and CC haplotypes). A−40T−37 is the only haplotype that could potentially bind to SMARCA3 RING finger. No affinity to TFs was predicted for the C−40T−37 configuration. The +18G allele could bind the ubiquitous GC-Box factor SP1 and the G triplet (G n≥3), and the allele T changes sequence affinity in favor of the NKX factor on the plus strand and 1B domain on the minus strand (Fig. 4BI). No TFBSs were predicted for the region when allele C represents the locus c.373+101. However, +101T could potentially create the TFBS to zinc factor AREB6 (Fig. 4BII). Nucleotide variations at +241, 243, 246, 249, and 259 loci define four different TFBSs patterns (Fig. 4BIII) uniting intronic haplotypes similar to their clustering in the phylogenetic tree (Fig. 3B). Hap 1, 4, 5, A, and E could potentially bind two modules of three and four TFs on the plus and the minus strands, respectively. Additionally, nucleotide G at +259 (Hap 1, 4, and A) creates TFBS to the EVII-myeloid transforming protein. This factor, together with FAC1 and ILF1, could be bound to the minus strand in Hap 2, 3, and B. The positive strand of the Hap 2, 3, and B could bind three TFs that are different from those predicted for Hap 1, 4, 5, A, and E. Specific patterns of TFBSs are expected also for Hap C and D. Overall, the enhancer binding factor ILF1 could be bound by the minus strand of all intronic haplotypes besides Hap D. The plus strand of the Hap 2, 3, B, and D could bind OCT1. Only Hap C and D have no affinity to FAC1. In contrast to other haplotypes, Hap 2, 3, and B possess binding capacity to the matrix associated SMARCA3 and could not bind the transcriptional enhancer TEAD and homeobox TF Nanog. Allele +323C (Fig. 4BIV) defines the minus strand affinity to MEIS1 and SMAD; however, allele T in this position generates new GATA1 site on the plus strand. Allele A +435A (Fig. 4BV), which was found to be monomorphic in LD, generates affinity to transcriptional repressor GF11, GATA1, and GTF2I control elements. Transition to +435C (AF393618) should be followed by loss of the mentioned and generation of the new affinity to zinc finger protein MYT1L.
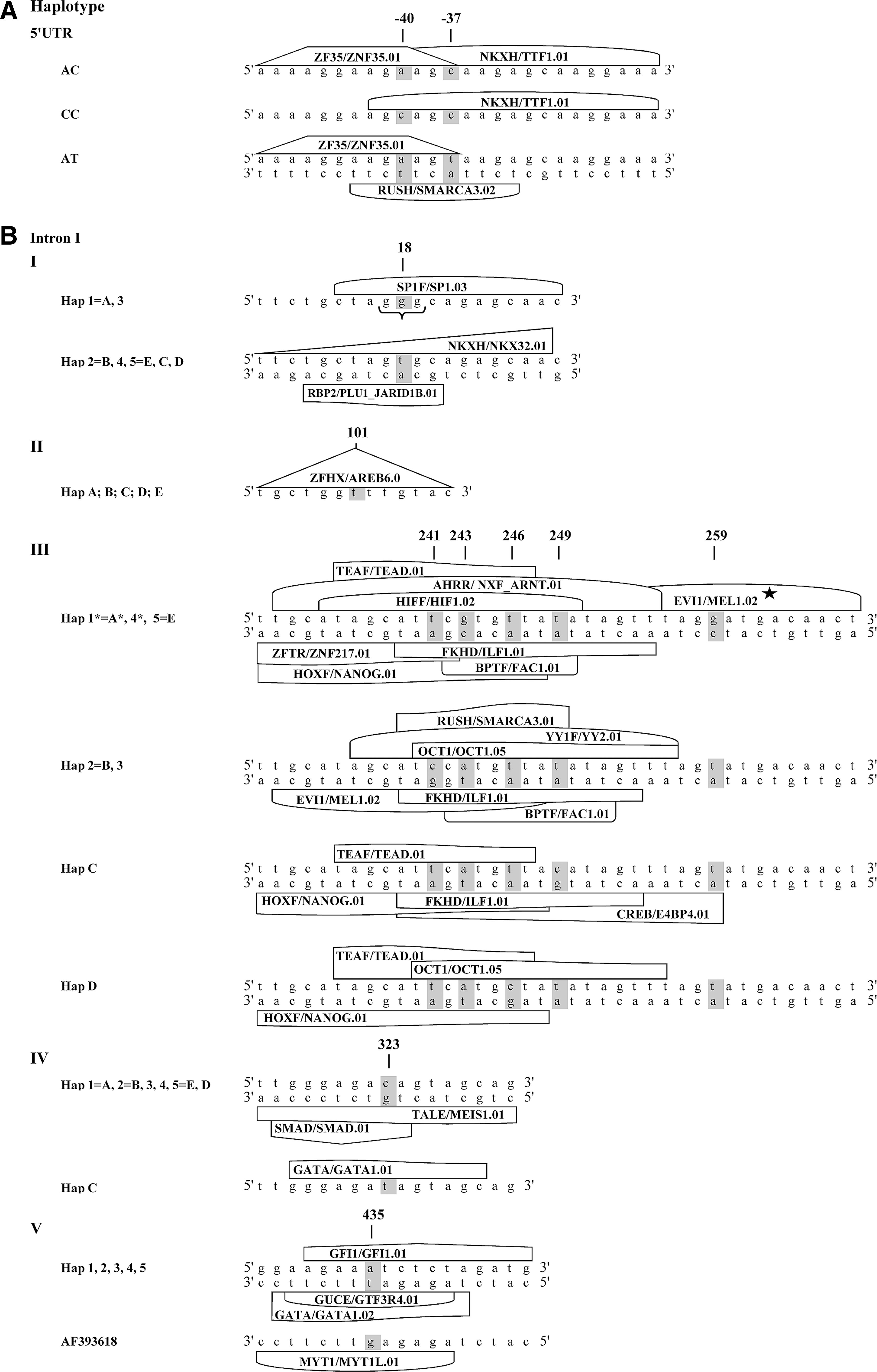
Haplotype-specific patterns of the transcription factor binding sites (TFBSs). (
Transcription factor family/matrix names are given for vertebrate according to the Genomatix database (
Targets for microRNA have not been predicted for any haplotype in the analyzed gene region.
Five CUG repeats of CTGCTA or CTGCTG motifs are located within the 190-bp region of intron I (Fig. 5A). Enrichment by CUG repeats in the 5′ region of intron I of the MSTN gene appears to be evolutionary conserved (Fig. 2B, C). One of the repeats is completely identical between sheep, goat, cattle, horse, swine, and humans. Two repeats are common between sheep, goat, and cattle. SNP c.373+18T>G is located one nucleotide downstream the first CUG repeat that is identical in sheep and goat and highly similar to related motifs in other species. Nucleotide transition from T to G could generate the additional hairpin and interhairpin boundary between the two CUG repeats that could finally lead to thermodynamically more stable pre-mRNA secondary structures (Fig. 5B, C).
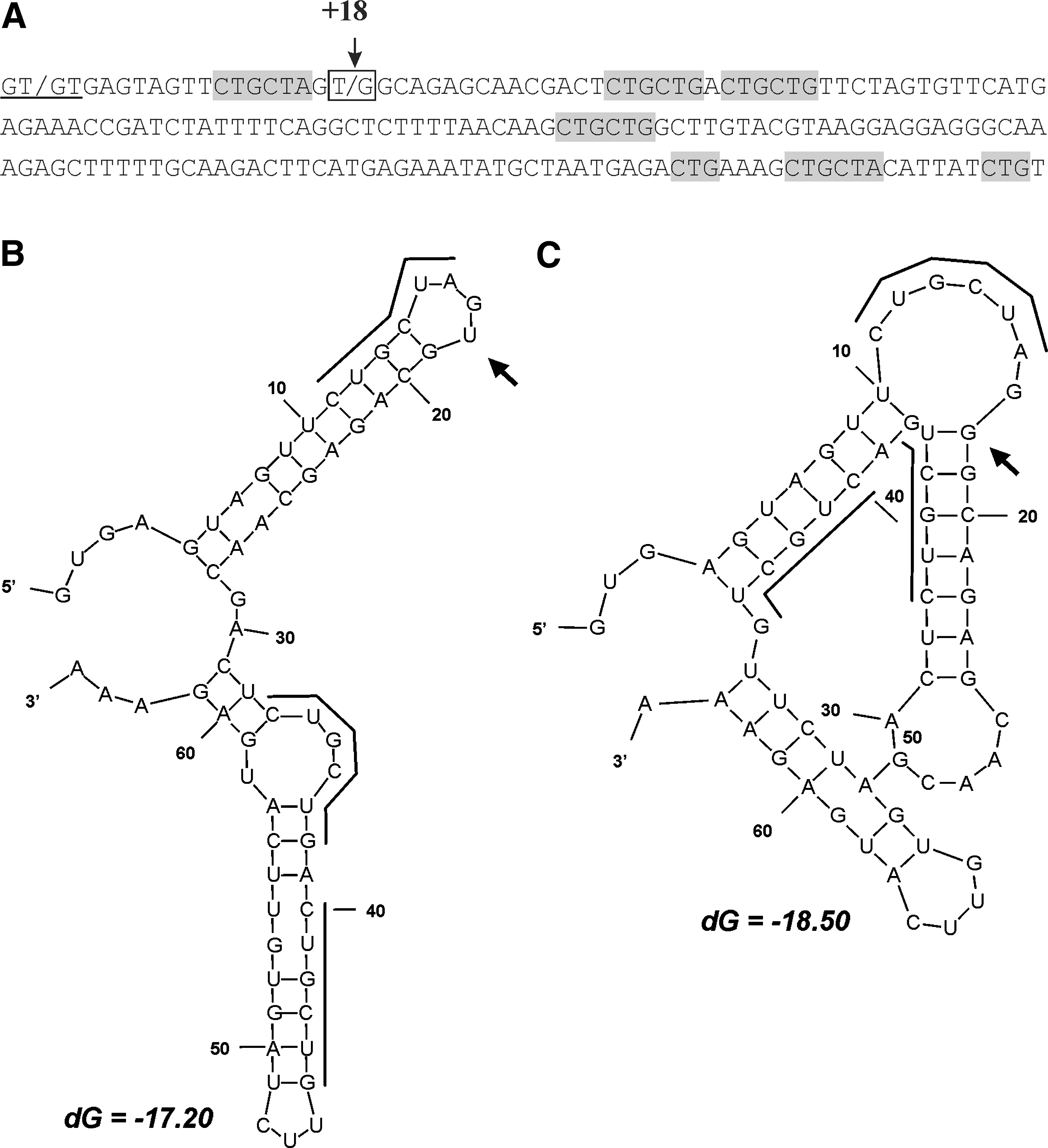
Impact of the SNP c.373+18 on the pre-mRNA secondary structure. (
DNA bendability was analyzed in silico for all SNPs and haplotype variants. DNA curvature was predicted only for the region around the +101 locus; if allele C is present, transition to allele T drastically decreases the sequence bendability (not shown). To prove the bent DNA experimentally, the mobility of the two amplicons of 645 bp and 270 bp (Fig. 1) was tested in agarose and PAAG gels (Fig. 6A and B, respectively). The 645-bp amplicon migrated in agarose gel accordingly to its size, but it was retarded as a 700-bp fragment in PAAG gel (55 bp retardation, R = 1.09) indicating the presence of bent DNA within the 645-bp fragment. No change in the mobility was observed in PAAG for the 270-bp fragment, indicating the absence of bent sites within the MS region.
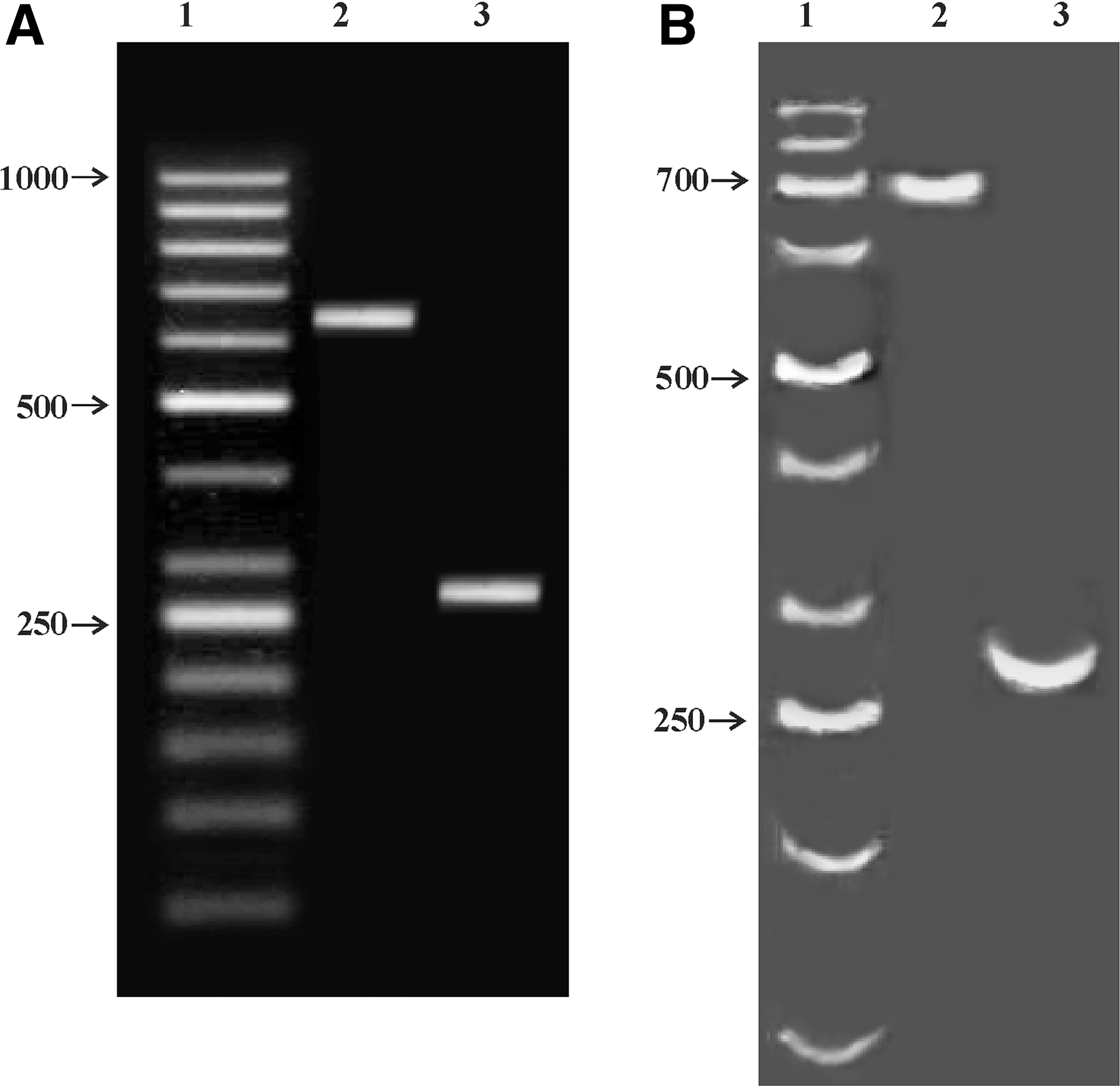
Experimental proof of the DNA curvature. (
Discussion
In the present study, we have analyzed the MSTN gene genetic diversity in the LD breed. The six loci were found to be polymorphic in the 1037-bp region encompassing the gene from the promoter to the MS locus of intron I. Two SNPs located in the 5′UTR at c.−40 and c.−37 positions are the same SNPs that were previously described by Clop et al. (2006); however, they were incorrectly indicated as c.−41 (Clop et al., 2006; Kijas et al., 2007) and c.−39 (Clop et al., 2006). Four intronic loci at c.373+18, +241, +243, and +259 were found to be polymorphic in LD. The other five loci at c.373+101, +240, +246, +249, and +323, previously described to be polymorphic in other breeds (Clop et al., 2006; Kijas et al., 2007; Hickford et al., 2010), were monomorphic in LD. The previously unidentified nucleotide at +434 (AF393618) and locus at c.373+435 were presented by allele A in all analyzed LD animals. The SNP c.373+435C > A is reported here for the first time and should be considered as a novel variation in the ovine MSTN gene.
Both the 5′UTR haplotype of C−40T−37 configuration, previously described for Texel (Clop et al., 2006), and Hap 1 of intron I were found to be predominant in LD. The intronic Hap 1, Hap 2, and Hap 5 observed in LD and the alleles A, B, and E identified by Hickford et al. (2010) in five breeds appear to be different only in one nucleotide at c.373+101. Both Hap 1 and allele A are obviously the most frequent haplotypes in LD and NZ Romney, respectively.
Hickford et al. (2010) found Hap A in all five breeds analyzed, and this was the only haplotype configuration observed in 19 animals of Texel. Texel was used in LD breeding programs to improve breed fertility and meat quality (
In our study, we have attempted to evaluate the possible impact in the gene function due to the variations in the noncoding gene region. To reach this goal, we constructed and compared the haplotype-specific TFBS patterns and analyzed the allele-specific DNA bendability and pre-mRNA secondary structures. The eventual effects of sequence variation in the gene function and phenotype manifestation will be discussed below.
The C−40T−37 haplotype of the 5′UTR appears not to possess strong affinity to any TFs. Kijas et al. (2007) revealed a significant negative effect of the c.−40C allele on forequarter weight and meat tenderness in ATex. Allele c.−40A is responsible for affinity to zinc finger protein ZNF35 and the A−40T−37 haplotype possesses affinity to SMARCA, the SWI/SNF-related chromatin remodeling transcriptional regulator of many muscle-specific genes (de la Serna et al., 2005; Simone, 2006; Zhou et al., 2009). The allele c.−37C defines the affinity to transcription termination factor TTF1 in both A−40C−37 and C−40C−37 haplotypes. Recently, it was shown that TTF1 could be involved in the downregulation of TGF-β target genes (Saito et al., 2009). The MSTN gene (GDF8) belongs to this family. One can speculate that the transcription of the MSTN gene would be less successful because of the TTF1 function and the extent of the myostatine negative effect on the muscle production could be decreased in the c.−37C animals compared with the c.−37T ones. This means that allele c.−37C is potentially beneficial, but allele c.−37T could produce a negative impact on meat production in sheep. Unfortunately, the SNP c.−37 has not yet been evaluated for its association with phenotype.
Nowadays, it is clear that introns are functionally active participants of gene and genome functionality. They encode intronic regulatory elements (Yutzey et al., 1989) participating in splicing, transcription, and recombination events including gene mobility. This means that not only conserved noncoding sequences possess functionality as regulatory elements, but also variable regions could also be adaptively active.
Similar to other mammals, exon I of the ovine MSTN gene ends with GT instead of the canonical AG. Change in the core donor site suggests potential for splicing errors and it is considered to be compensatory regulated by intronic splicing enhancers or silencers (Wang and Burge, 2008). There are several reasons that suggest the c.373+18G>T locus is functionally active with respect to splicing. Allele +18G creates the binding site on SP1, a ubiquitous stimulating protein, and the G triplet (G n≥3), which is a well-characterized evolutionary conserved intronic splicing enhancer (Yeo et al., 2004) and can enhance exon inclusion (McCullough and Berget, 1997). In contrast, the allele +18T creates sequence affinity for the NKX32 transcriptional repressor (Champigny et al., 2009) and one of the RBP2 factors that can also repress transcription through demethylation (Christensen et al., 2007). The 5′ region of intron I is enriched by the CUG repeats known as splicing regulatory motifs recognized by different regulatory proteins similar to muscleblind-like (MBNL) proteins (Warf and Berling, 2007). The SNP +18G>T does not change the CUG repeats pattern but could influence pre-mRNA secondary structure, an integral part of splice site recognition (Hiller et al., 2007). In our study, we found that nucleotide G at +18 position initiates single strandedness in the first CUG repeat and G triplet location. Formation of an additional hairpin in the region could be followed by reorganization of the spatial topology between the two other CUG repeats. Such perturbation of the pre-mRNA secondary structure could potentially influence sequence interaction with different regulatory proteins and the efficiency in the lariat formation (Chasin, 2007; Hiller et al., 2007; Aznarez et al., 2008; Wang and Burge, 2008). Altogether, the affinity to SP1, G triplet appearance, higher single strandedness of the region, and pre-mRNA secondary structure perturbation could potentially increase the transcription and splicing efficiency of the +18G sequence variant compared to the +18T allele. Consequently, the +18T variant should be considered as advantageous in meat breeding programs in sheep. Our suggestions are in good agreement with data obtained in NZ Romney sheep on the association between the Hap B (+18T) and A (+18G) and the increased loin yield and the decreased leg, loin, and total yield of lean meat, respectively (Hickford et al., 2010).
The SNP at locus c.373+101 is the only variation discriminating the Hap 1, 2, and 5 observed in our study from the Hap A, B, and E described by Hickford et al. (2010). The T nucleotide at +101 creates the transcription repressor AREB6 binding site. The C nucleotide could change sequence bendability following the formation of the DNA curvature. Curved DNA potentially could be more beneficial for sequence interaction with intronic regulatory elements in replication, transcription, repair, nucleosome formation, and recombination processes (Lobov et al., 2001; Rothenburg et al., 2001; Yamamura and Nomura, 2001; Li et al., 2003; Gimenes et al., 2009). In our study, the high probability of recombination was predicted exactly for the DNA region around the +101 SNP.
Four TFBS patterns were generated when the nucleotide transitions at +241, 243, 246, 249, and 259 loci were analyzed together. The distribution of the individual haplotypes between TFBS patterns was similar to haplotype clustering in the phylogenetic tree. Comparison of the putative functionality between Hap 1 = A and Hap 2 = B appears to be the most interesting as the decrease and increase of the meat performance traits are associated with Hap A and Hap B, respectively (Hickford et al., 2010). In fact, the Hap 1 = A sequence variant could specifically bind those TFs that potentially negatively influence myogenesis. These include TEAD, a modulator of slow muscle gene expression (Tsika et al., 2008), NXF, a transcription activator (Ooe et al., 2004), HIF1, a factor involved in the upregulation of several target genes in skeletal muscle (Kubis et al., 2005), and Nanog, a suppressor of the myogenic differentiation program (Lang et al., 2009). In contrast, the Hap 2 = B sequence could specifically bind OCT1, which might potentially function similarly to OCT4, another octamer binding protein found to be a transcriptional repressor during muscle terminal differentiation (Lang et al., 2009).
The nucleotide C at the c.373+323 locus in most haplotypes creates the binding sites on the minus strand to both SMAD proteins, involved in muscle mass regulation in adulthood (Sartori et al., 2009), and the MEIS1 cofactor of TALE (Hox) family, critical for many aspects of animal morphogenesis (reviewed by Mann et al., 2009). TALE family cofactors increase the size of TFBS and help to impose additional structure onto otherwise unstructured homeodomain and nonhomeodomain residues, allowing them to read additional features present in particular TFBSs (Mann et al., 2009). This may have a special role in the sequence interaction with other regulatory proteins, particularly with SMAD proteins. The nucleotide T at +323 defines the affinity to the nonspecific transcriptional activator of the GATA family involved in the muscle growth regulation through the calcineurin signaling pathways (Michel et al., 2004).
Nucleotide A at the c.373+435 locus creates sites binding for transcriptional repressor CFI1 and general transcription factor GTF3, which both participate in the TGF-β–mediated signaling pathways (Chimge et al., 2008; Katoh and Katoh, 2010).
The majority of the described TFs and TFBSs are derived from human/mouse data and we have only assumed that TFs could work similarly in sheep. Some of our assumptions might be tested in the future by using the myoblast/MSTN reporter system described by Du et al. (2007).
Overall, our findings suggest that the SNPs analyzed in this study may be significant in the sheep MSTN gene function. It can be also concluded that in silico analysis has become an indispensable tool in modern molecular genetics analysis and multiple genotyping data evaluation.
Footnotes
Acknowledgments
This study was funded by the Latvian Council of Science (Grant Nos. 08.2190, 09.1368, and 09.1550). The authors thank the Latvian Ministry of Agriculture for additional financial support (Contract Nos. 120706/S386 and 201006/C-266). Collaboration between institutions involved was additionally supported by the 2008–2011 NordPlus Horizontal Network cooperation “Implantation of genomics and bioinformatics in agricultural practice.” The authors are grateful to Prof. N. Sjakste for reading the manuscript and helpful discussions.
Disclosure Statement
No competing financial interests exist.