
Abstract
The NV epitope, a dominant helper determinant from the circumsporozoite antigen of Plasmodium falciparum, is strongly immunogenic and can provide help for cytotoxic T-lymphocyte (CTL) activation. In this study, we evaluated whether the addition of NV peptide can augment the efficacy of peptide-pulsed dendritic cell (DC) immunization in vivo. Using B16 melanoma as tumor model, we demonstrated that DCs pulsed with both NV and gp100 (a melanoma-specific antigen) peptide enhanced immune priming and protection from tumor challenge in vivo. Further, we showed the mechanisms of the NV epitope that help CTL activation; MHC-II–restricted NV peptide induced dramatically more effective helper cells, with a higher level of CD40L expression and IFN-γ production, which, in turn, more effectively conditioned DCs for CTL activation. The improved helper cells also induced greater IL-12 production by DCs, accounting for the reciprocal T-helper polarization to Th1, and increased the expression of costimulatory molecules. Collectively, these findings demonstrate that NV peptide in addition to tumor antigen–pulsed DC immunizations augment helper cell activation, which in turn promotes maturation of DC, and enhance in vivo antitumor activity.
Introduction
The NV epitope (NANP-NVDP-NANP), a dominant helper determinant from the circumsporozoite antigen of the human malaria parasite Plasmodium falciparum, is an agent capable of stimulating a potent T-helper response. Gerloni et al. demonstrated that immunization of B cells transfected by γ1 plasmid with expression of NP66–72, a CTL epitope from influenza viral A, and NV epitope in linked association in transgenic Ig (antigenized antibody) produce more qualitative and quantitative effect on the CTL response compared with the plasmid expressing only NP epitope in transgenic Ig (Langlade-Demoyen et al., 2003; Rizzi and Castiglioni, 2004). In the same DNA immunization system, they also found that NV epitope, serving as a strongly immunogenic Th-cell determinant, can pave the way for the response to the otherwise nonimmunogenic cryptic Th-cell determinant and B-cell epitope as well (Gerloni et al., 1999, 2000).
Dendritic cells (DCs) are potent professional antigen-presenting cells, which interact with T cells and initiate their responses. The antigen-presenting capability of DCs makes them attractive vehicles for the delivery of therapeutic peptide cancer vaccines (Steinman, 1991; Chauvin and Josien, 2009). In the present study, we evaluated the capacity of NV peptide to enhance DC-based tumor vaccines in the setting of poorly immunogenic B16 melanoma and its subline (D5). We demonstrate that simultaneous pulsing of DCs with NV peptide and gp100, one of the best-characterized melanoma-associated antigens, results in pronounced enhancement of vaccine-mediated immune priming and therapeutic efficacy in vivo. Further, we investigated the possible mechanisms by which NV peptide augments class I peptide–restricted CTL response. NV peptide can activate T-helper cells to a higher level compared with control peptide. Using a well-characterized in vitro system, we further demonstrated that the interaction between NV-pulsed DCs and CD4+ T cells results in upregulation of costimulators on DCs and increased expression of IL-12, leading to a qualitative as well as quantitative augmentation in CTL immune response.
Materials and Methods
Animals
Six-to-eight-week-old female C57BL/6 mice were purchased from the Laboratory Animal Center of Chinese Academy of Science and housed in the pathogen-free animal facility of Fudan University.
Peptides, cell lines, antibodies, and recombinant cytokines
gp100, a melanoma-associated antigen, is expressed on normal melanocytes and B16 melanoma. The MHC class I–restricted gp10025–33 peptide (KVPR-NQDW-L) and the mouse MHC class II–restricted NV peptide (NANP-NVDP-NANP) derived from the circumsporozoite antigen of the human malaria parasite Plasmodium falciparum were synthesized by Biotec Company. The MHC class II–restricted OVA peptide (OVA323–339, ISRA-VHAA-HAEI-ENEA-GR) used as irrelevant control peptides were also synthesized by Biotec Company. All of the peptides were made by standard 9-fluorenylmethoxycarbonyl chemistry and purified by reverse-phase high-performance liquid chromatography, and purity of >80% was confirmed by mass spectrometry. B16F10 melanoma characterized by poor immunogenicity and high metastasis is a subclone of the B16 tumor (denoted F10). Cell lines were cultured in a medium of RPMI 1640 (GIBIO) supplemented with 10% fetal calf serum (FBS; Gibco) and 0.2% penicillin/streptomycin. We obtained the following antibodies commercially: FITC-labeled rat anti-mouse CD8α, PE-labeled rat anti-mouse IFN-γ, FITC-labeled rat anti-mouse CD80, CD86, and H-2Db, PE-labeled rat anti-mouse CD11c, and PE-labeled rat anti-mouse CD40 and CD40L. All antibodies were purchased from BD PharMingen. Recombinant murine granulocyte macrophage colony-stimulating factor (GM-CSF) with a specific activity of >5 × 106 units/mg were used to generate DCs. Recombinant human IL-2 with a specific activity of 18 × 106 IU/mg was used to culture T cells. All recombinant cytokines were purchased from Peprotech, Inc.
Generation of bone marrow–derived DCs
Bone marrow–derived murine DCs were generated as previously described (Hanada et al., 2005). Bone marrow cells from the femur and tibiae of C57BL/6 mice were grown at a starting concentration of 1 × 106 cells/mL in RPMI 1640 supplemented with 10% fetal bovine serum,
Antigen pulsing of DCs
On day 7, DCs were incubated at 37°C in 5% CO2 with gp100 peptide or NV peptide (denoted DC-gp100 or DC-NV) at 10 μg/mL for 3 h. After 3 h incubation, DCs were harvested, washed twice in HBSS (Life Technologies, Inc.), and resuspended in HBSS for additional studies. In parallel experiments, DCs were co-incubated with gp100 peptide in the presence of 10 μg/mL NV peptide (denoted DC(NV)-gp100).
Immunizations and tumor challenge
We subcutaneously injected 2 × 106 DC(NV)-gp100 or the same number of DC, DC-NV, or DC-gp100 into the right flank region of C57BL/6 mice, and the same immunization was repeated twice at 1-week intervals. Tumor challenge was initiated by subcutaneously injecting 5 × 104 viable B16F10 melanoma cells into the rear leg of the immunized mice at 1 week after the final immunization. Tumor occurrence was observed every other day. The length and width of tumor mass were measured with a caliper every other day, and tumor area was expressed as length × width. When the tumor size could not be measured by a caliper, we denoted the tumor size as zero. Five mice in each group were observed for their survival period. Mice that became moribund were killed. All experiments were performed three times using individual treatment groups of five mice. Data are representative of three experiments.
Proliferation assays
Spleen cells from naive C57BL/6 mice were harvested, and cell suspensions placed on the top of a column full of nylon wool incubated. After about 20 min, the column was washed using 10 mL complete culture medium to enrich for T cells. The purity of the enriched T cells harvested was determined by FACS using FITC-labeled anti-CD3 monoclone antibody. The enriched T cells (about 90% purity) were used as responder cells. The DCs pulsed with NV peptide or OVA peptide were inactivated with 50 μg/mL mitomycin for 30 min and then used as antigen-presenting cells. Five hundred thousand responder cells and the inactivated DCs (5 × 104 cells) were cultured on a flat-bottomed 96-well microtiter plate (Corning) for 7 days in the presence of 20 U/mL recombinant human IL-2. During the last 18 h of the culture, 1 μCi 3H-thymidine (Shanghai Atomic Energy Institute, Chinese Academy of Science) was added to each well. 3H-thymidine incorporation was measured in a liquid scintillation counter (Shanghai Atomic Energy Institute, Chinese Academy of Science). Results are expressed as a stimulation index (SI) calculated as the following ratio: (cpm of cells cultured in the presence of synthetic peptide)/(cpm of cells cultured in the absence of peptide). All samples were assayed in triplicate and the values are shown as the means ± SD.
Assay of cytokine production
Seven days after final immunization, the nonadherent splenocytes derived from immunized mice were harvested, and cell suspensions passed through nylon wool to purify T cells as previously described. Then, 5 × 105 enriched primed T cells (about 90% purity) derived from mice immunized with NV or OVA peptide were cocultured with NV or OVA peptide–loaded DCs (5 × 105cells) on 24-well microtiter plates (Corning) for 36 h. The culture supernatant (100 μL) was tested for IL-12 and IFN-γ production. The amounts of cytokines were determined using a standard sandwich ELISA technique with corresponding kits from BD PharMingen.
Cytotoxicity assay
Splenocytes from immunized mice were harvested and restimulated in vitro with 10 μg/mL gp100 peptide in presence of 20 U/mL IL-2 for 7 days. The cultured cells served as effector cells and were then tested for tumor-specific cytotoxicity with a flow cytometric CFSE/PI cytotoxicity assay as previously described (Lecoeur et al., 2001; Chu et al., 2006). Briefly, effector cells (106) were labeled with 200 nM CFSE in PBS for 15 min at 37°C in a volume of 1 mL. After washing in PBS, CFSE-labeled effector cells were seeded with 2 × 104 target cells at different E:T ratios of 10:1, 20:1, 40:1, and 80:1. The cells were incubated in 96-well microplates in a total volume of 200 μL complete medium for 4 h in a 5% CO2 incubator at 37°C. The cell mixtures were then washed in PBS–1% BSA containing 0.1% sodium azide (NaN3) and incubated in 200 μL of the same buffer containing 20 mg/mL PI for 20 min at 4°C in the dark. The cell samples were then washed and fixed with 1% paraformaldehyde for 20 min. Acquisition was performed right afterward on a FACScan flow cytometer. Flow cytometry analysis was performed in three steps: (1) selection of target cells on the FSC/CFSE dot plot, distinguishable from effectors cells by their CFSE negativity; (2) gating and discarding of apoptotic bodies/debris according to their low cell size (FSClo)—weak PI incorporation (PIlo); and (3) determination of the percentage of target lysis, corresponding to the percentage of apoptotic (PIlo + PIhi) cells (sample lysis). In parallel, basal lysis was quantified, corresponding to the percentage of apoptotic target cells in the absence of effector cells. The percentage of specific lysis for a given E:T ratio was calculated using the following formula: 100 × (% sample lysis −% basal lysis)/100 −% basal lysis. For each E:T ratio, 10,000 target cells were acquired.
Ex vivo intracellular IFN-γ staining assay
Splenocytes from immunized mice were incubated for 5 h at 5% CO2/37°C in 96-U-well plates, at 5 × 105 cells/well, along with 20 U/mL rIL-2, 10 μg/mL gp100 peptide, and 1 μL/well Golgi plug (BD PharMingen). Samples were blocked with 2% decomplemented fetal bovine serum (15 min, 4°C) before labeling with 0.5 μg/well FITC-anti-mouse CD8α. Following surface labeling, the cells were fixed with 1% formaldehyde (20 min, 4°C) and then permeabilized with 0.5% saponin (10 min, 4°C) before intracellular labeling with 0.5 μg/well PE rat anti-mouse IFN-γ for 20 min at 4°C. After a final wash, the cells were resuspended in PBS and analyzed immediately by FACS Calibur using CellQuest software (BD Biosciences).
Immunotherapy of the preestablished B16 melanoma model
For immunotherapy of established melanoma, tumor-bearing mice were established by subcutaneous inoculation of 5 × 104 B16F10 melanoma cells. DC(NV)-gp100 or control suspensions were subcutaneously injected into the flank region of the tumor-bearing mice at 6 days after tumor inoculation. The same immunization was repeated twice every 3 days. Each group contained five mice. The length and width of the tumor mass were measured with a caliper every other day after tumor inoculation. Tumor area was expressed as length × width. The tumor-bearing mice were observed for their survival period.
FACS analysis
Splenic T cells (5 × 105) from mice immunized with NV or OVA peptide were incubated in 24-well plates with 5 × 105 NV or OVA peptide–loaded DCs for 36 h. T cell–DC conjugates were disrupted by washing with HBSS with 1% BSA (Huamei Company), 25 mM HEPES (Sigma), and 15 mM EDTA (Sigma). PE-conjugated rat anti-mouse CD40L and FITC-labeled rat anti-mouse CD4 were initially added to examine for CD40L expression on CD4+ T cells by FACScan (Becton Dickinson Immunocytometry Systems). The phenotypic analysis of DCs was performed by FACScan using the following phycoerythrin or fluorescein isothiocyanate monoclonal antibodies: PE-conjugated rat anti-mouse CD40, FITC-labeled rat anti-mouse CD80, CD86, and H-2Kb.
Statistical analysis
The statistical analyses to compare tumor size, T-cell proliferation, CTL cytotoxicity, and cytokine production between treatment and control groups were determined using the Mann–Whitney nonparametric U test. Comparisons of mouse survival between treatment and control groups were determined with a Kaplan–Meier test.
Results
More effective elicitation of protective antitumor immunity by immunization with DC concomitantly pulsed with NV and Gp100
To determine whether introduction of NV epitope into a DC-based vaccine can enhance immune priming of mice to the relative weak immunogenic melanoma-associated antigen gp100, B6 mice were immunized with DC-gp100 with or without concomitant pulsing with NV peptide. Seven days after the last immunization, these mice were challenged with B16 melanoma cells. As shown in Figure 1, mice receiving DC-gp100 without being pulsed with NV only obtained partial protection from B16 melanoma challenge, only 33% of mice were tumor free. The mean survival time of these mice was 52 days. However, a significant increase of tumor-free survival and inhibition of melanoma growth were observed following immunization of mice with DC-gp100 and NV peptide (mean survival time: 256 days vs. 52 days; p = 0.0013). Mice immunized with either DC only or DC pulsed with NV only had no apparent protective efficacy. The surviving mice immunized with DC-gp100 and NV peptide were rechallenged at 60 days after tumor inoculation with 1 × 105 D5 melanoma cells (i.e., a twofold higher challenge dose); all showed complete protection (Table 1). An additional cohort of the survival mice were challenged with irrelevant EL-4 tumor cells and showed no protection to this unrelated tumor (data not shown). These findings indicated that NV epitope improved the antitumor efficacy of DC-gp100.

Protective antitumor immunity by immunization with the DC(NV)-gp100 vaccine. C57BL/6 mice were subcutaneously immunized with 2 × 106 DC(NV)-gp100 at 21, 14, and 7 days before challenge with B16 melanoma cells. Control groups of mice were immunized with either DC-gp100, DC-NV, or DC.
All cured mice were rechallenged at 60 days after tumor inoculation with 5 × 104 B16 melanoma cells. This table shows the faction of survival (survivor/total) after rechallenge with B16F10.
DC, dendritic cell.
More potent induction of therapeutic antitumor immunity by immunization with DCs concomitantly pulsed with NV and Gp100
DC (NV)-gp100 immunizations protect mice from subsequent tumor challenge. To evaluate this vaccination strategy in a model more analogous to a clinical setting, the therapeutic effect of DC(NV)-gp100 immunizations was tested in mice with preestablished tumors. In a 6-day treatment model, B6 mice harboring preestablished tumors were treated with DCs, DC(NV), DC-gp100, and DC(NV)-gp100, respectively, three times at an interval of 4 days. Figure 2A shows the inhibition of tumor growth in the tumor-bearing mice immunized with various vaccines. DC-gp100 vaccines inhibited tumor growth and prolonged the mean survival time moderately (mean = 38 days, compared with control group; p < 0.05). However, treatment with DC(NV)-gp100 resulted in a significant delay in tumor growth and potent prolongation of mean survival time (mean = 55 days, compared with DC vaccine or NV pulsing-DC alone; p < 0.01). These results suggested that NV enhanced the therapeutic efficacy of DC immunizations as well as protective immunity against a poorly immunogenic tumor.
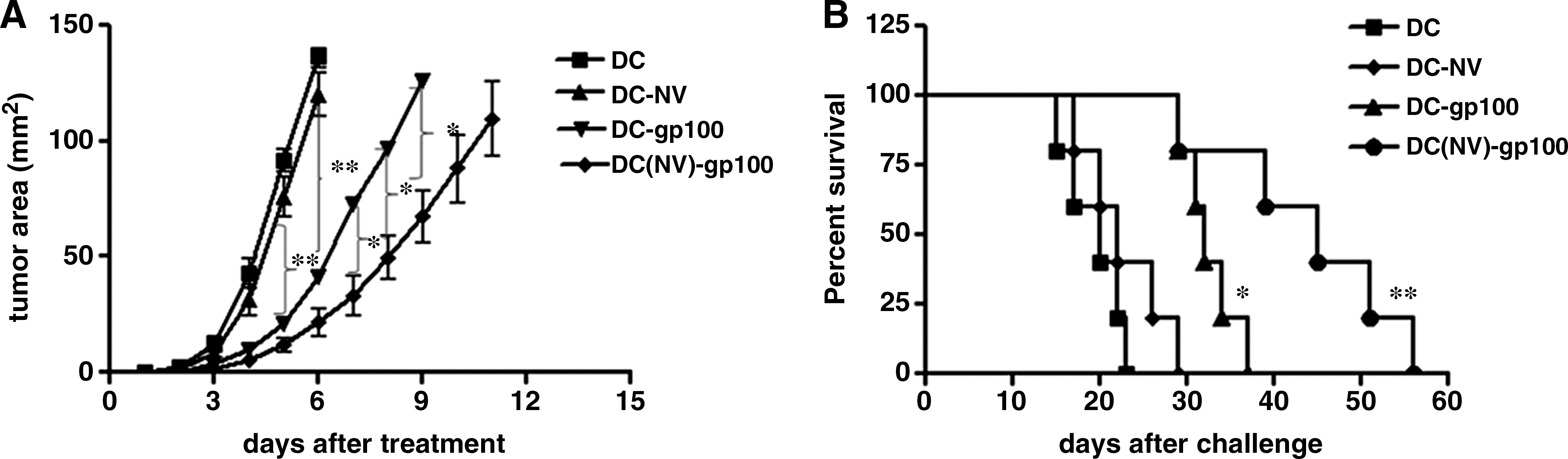
Inhibition of tumor growth in tumor-bearing mice by immunizations with DC(NV)-gp100 vaccine. C57BL/6 mice were subcutaneously injected with 5 × 104 B16F10 melanoma. Six days later, tumor-bearing mice were immunized with 2 × 106 DC(NV)-gp100, DC-gp100, DC-NV, or DC.
Enhanced CTL response induced by immunization with DCs concomitantly pulsed with NV and gp100
The splenocytes derived from the tumor-bearing mice immunized with various vaccines were restimulated in vitro with inactivated spelenocytes pulsed with gp100 peptide. The results in Figure 3 are representative of multiple experiments showing that when the mice immunized with DCs concomitantly pulsed with NV and gp100, the frequency of gp100-specific CD8+ CTL was 1.87% as determined by intracellular staining, which is far greater than DC vaccine or NV pulsing-DC alone and twofold higher than DC-gp100 vaccination. Also, the ability of the IFN-γ–producing CD8+ T cells to kill target cells was then tested following a 6-day expansion with peptide-loaded APCs in vitro. Consistent with the induction of IFN-γ–producing CD8+ T cells, as shown in Figure 4, highest CTL response was induced in the mice immunized with DC(NV)-gp100 vaccine, and these mice exhibited tumor-specific splenic CTL response potently (compared with control group; p < 0.01). A certain level of CTL could be induced in mice immunized by DC-gp100 vaccine (compared with control group; p < 0.05), but no significant CTL induction was observed in mice immunized with DC-NV. In determining CTL activity, no cytotoxicity on syngeneic EL-4 T lymphoma cells was found with these induced lymphocytes (data not shown). These results demonstrated that NV epitope could enhance the ability of DC-gp100 to expand the precursor of gp100-specific CTL and augment these gp100-specific CTLs to kill the target melanoma cell.

Immunization of mice with DC(NV)-gp100 boosts the frequency of gp100-specific CD8+ T cells. C57BL/6 mice were vaccinated with DC

gp100-specific CTL activity after immunization with DC(NV)-gp100. One week after the final immunization, splenic lymphocytes from the mice immunized with DC(NV)-gp100, DC-gp100, DC-NV, or DC were cocultured with inactivated B16 cells for 7 days in the presence of recombinant murine IL-2 (20 U/mL) and then collected. The B16 melanoma cells were used as target cells. CTL cytotoxicity was determined at E:T ratios of 10:1, 20:1, 40:1, and 80:1 by double staining of CFSE and PI. Each of the curves shown represents the mean percentage of specific lysis of CTLs induced by various immunizations. Standard deviation is indicated as bars within the graph. *p-value < 0.05; **p-value < 0.01. CTL, cytotoxic T lymphocyte.
Optimum CD4+ T-cell proliferative response was achieved by stimulation with NV peptide–loaded DCs
As previously described, a potent T-helper-cell response can enhance CTL reactivity to tumors (Ahlers et al., 2001; Kianizad et al., 2007), and so initial experiments were designed to analyze the ability of NV peptide–loaded DCs to activate CD4+ T cells. T lymphocytes from untreated mice were cultured for 7 days under stimulatory DCs, NV peptide alone, and DCs pulsed with NV or OVA peptide. As evident in Figure 5A, free NV peptide or DCs alone induced only weak cell proliferation when added to purified T cells. Importantly, however, when DCs were loaded with NV peptide prior to their culture with responder T lymphocytes, a dramatic increase of the proliferative response was observed. Further, NV peptide–preloaded DCs elicited a 1.8-fold higher response than OVA peptide–loaded DCs. IFN-γ production by host-derived T cells and CD40L expression on T cells correlate with maturation of DCs, which in turn could activate CTLs (Schoenberger et al., 1998; Kalinski et al., 2006). We next examined the IFN-γ production by ELISA (Fig. 5B) and CD40L expression by FACS (Fig. 5C) after T lymphocytes from NV or OVA peptide–primed mice were cultured for 36 h under restimulation with DCs pulsed with NV or OVA peptide, respectively. In three independent experiments, NV peptide–primed T cells cocultured with NV-loaded DCs secreted greater amounts of IFN-γ than control OVA peptide at all graded doses of peptide (0.01–100 μg/mL). When the concentration of pulsed NV peptide is 1 μg/mL, the IFN-γ production (457 pg/mL) was fourfold greater than control peptide. At the highest dose tested (100 μg/mL), the frequency of CD4+ T-cell–expressed CD40L among all of primed T cells following stimulation with NV peptide–loaded DCs was also greater than OVA peptide (7.52% vs. 3.68%). These data indicated that NV peptide triggered the activation of CD4+ T cells and shifts the quality of the response toward Th1 more than OVA peptide.

Differential upregulation of costimulators on DCs by NV epitope
The maturation of DCs is essential for the activation of CTLs. Previous studies have shown that costimulators such as CD40-CD40L and B7(B7.1, B7.2)-CD28 play a vital role in the reciprocal activation of DCs and T cells when they interacted (Bennett et al., 1998; López-Bravo and Ardavín, 2008). We also determined whether the ability of NV epitope to enhance immune response to tumor antigen gp100 resulted from an upregulation of costimulatory molecules on the APCs during interaction between CD4+ T cell and NV-pulsed DCs. Bone marrow–derived DCs were loaded with peptide OVA or peptide NV followed by incubation with 5 × 105 primed T cells from mice immunized with NV or OVA peptide previously for 36 h. Then, the cells were gated on class II or CD11c+ and examined for costimulatory molecules. As evident in Figure 6, compared with control peptide, MFI of B7-1, B7-2, MHC-I, and CD40 on DCs increased statistically in the NV peptide group (p < 0.05), indicating the ability of NV peptide to induce more expression of B7-1, B7-2, MHC-I, and CD40 on DCs and greater maturation of DCs than control peptide OVA.
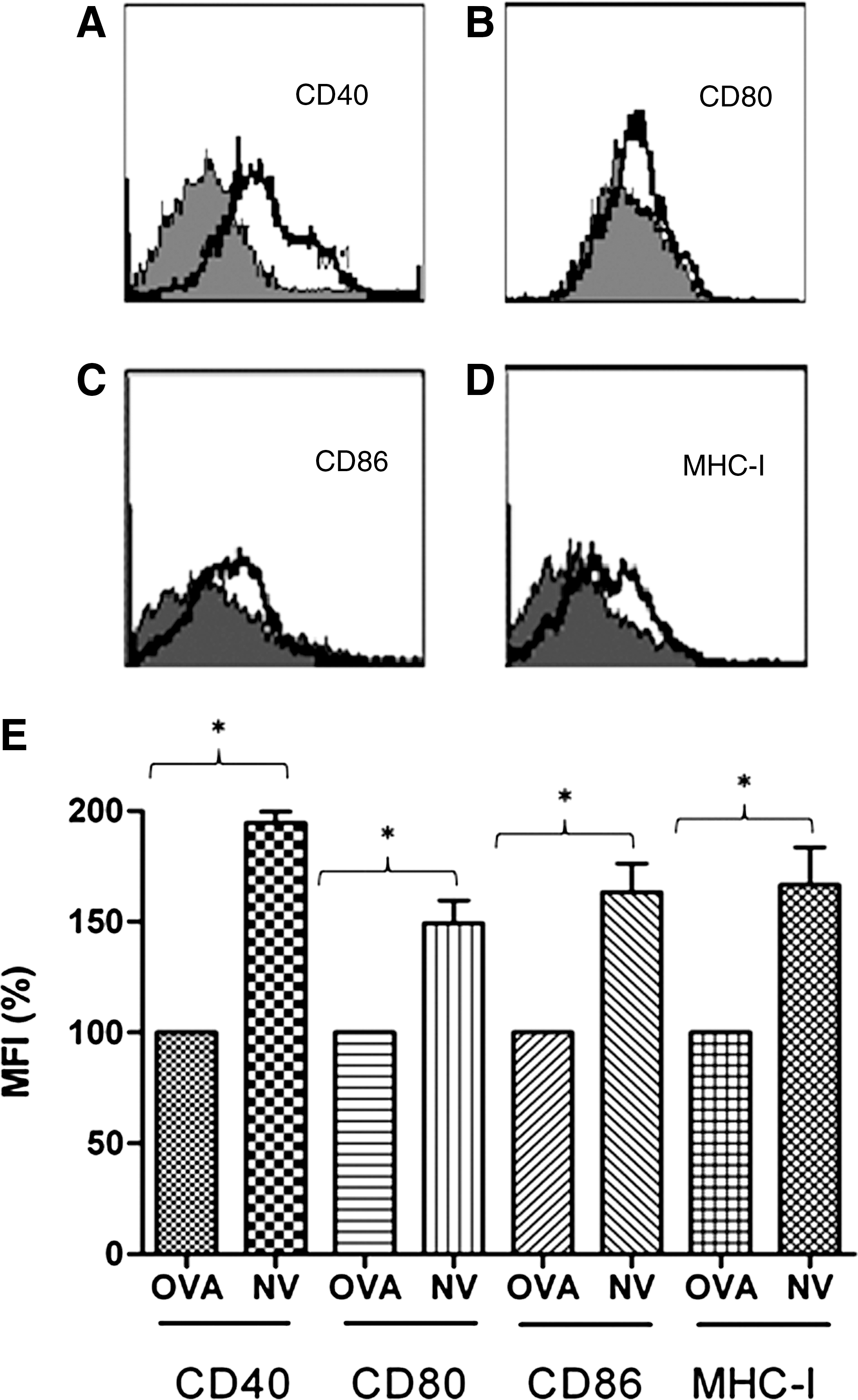
Differential expression of costimulatory molecules on DCs. NV- or OVA-loaded DCs (5 × 105cells/well of 24-well plate) grown with GM-CSF (50 ng/mL) were cocultured for 36 h with 5 × 105 primed T cells from mice immunized with NV or OVA peptide previously. Then, the cells were harvested for phenotypic analysis of DCs. Gated CD11c+ cells were examined for costimulatory molecules CD40
Interaction of NV-pulsed DCs and T cells could increase IL-12 production
IL-12 production by DCs could augment the function of CTLs (Giorgio, 2003). To clarify whether enhancement of immune response to gp100 triggered by NV epitope is related with its ability to induce more production of IL-12 by DCs, ELISA was performed to detect the level of IL-12 after interaction between T cell and NV-pulsed DCs in vitro. From Figure 7, we can see that, compared with control peptide, coculture of NV peptide–pulsed DCs with T cells can produce more IL-12 in the culture supernatant. With elevated concentration of pulsed NV peptide, the production of IL-12 is also increased. When the concentration of pulsed NV peptide is 10 μg/mL, the concentration of IL-12 in supernatant can reach 752 pg/mL, which was augmented >2-fold by coculture of control peptide–pulsed DCs with T cells.

Production of IL-12 by DCs is dramatically increased when cocultured with T cells in the presence of NV peptide. NV- or OVA-loaded DCs (5 × 105cells/well of a 24-well plate) grown with GM-CSF (50 ng/mL) were cocultured for 36 h with 5 × 105 primed T cells from mice immunized with NV or OVA peptide previously. Thirty-six hours later, culture supernatants were assayed for IL-12 by ELISA. These results were highly reproducible in three separate experiments. Each of the curves shown represents the mean concentration of IL-12 in supernatant. The standard deviation is indicated as bars within the graph. *p-value < 0.05; **p-value < 0.01.
Discussion
The cytolytic T-cell response is believed to be critical in mediating potential antitumor immunity (Wang and Rosenberg, 1999). Thus, studies of tumor immunity have primarily focused on the identification of MHC class I epitopes for tumor-associated antigens such as MAGE-1 (Sun et al., 2002), HER-2/neu (Kim et al., 2008; Mittendorf et al., 2008), tyrosinase (Pavelko et al., 2010), and gp-100 (Ly et al., 2010). However, several clinical trials assessing the feasibility and efficacy of cancer vaccination with peptide-based MHC class I peptides demonstrated that the effect of these vaccines are limited (Bronte and Mocellin, 2009). Several problems have been identified with MHC class I peptide vaccination, including the inability to generate peptide-specific precursors that directly recognize naturally processed antigen, to produce a significant precursor frequency and to overcome the threshold required for recognition by tumor-specific T cells in vivo. Thus, expanding peptide-specific precursors that directly recognize naturally processed tumor antigen is essential for the development of effective tumor vaccine.
Several studies have shown that CD4+ helper T cells are essential for the CD8+ cytotoxic T-cell response to pathogens and tumor cells (Bennett et al., 2001; Wang and Livingstone, 2003; Smith et al., 2004; Savelyeva et al., 2005). The memory CD8+ T cells that are generated without CD4 help are defective in their ability to respond to secondary encounters with antigen (Shedlock and Shen, 2003; Sun and Beven, 2003; Bevan, 2004; Sun et al., 2004). These studies also show that a higher-affinity helper epitope enhances peptide-based vaccine, induces more CTLs, skews helper cells toward Th1 cytokine production, and protects against viral and tumor challenge (Ahlers et al., 2001; Kianizad et al., 2007). Immunization strategies combining tumor antigen MHC class I epitopes with universally recognized MHC class II epitopes such as PADRE (Alexander et al., 1998), the promiscuous epitopes of tetanus toxoid (Castellino et al., 2009), and HBV helper epitope (Bayard et al., 2010) showed that not only immunity to the helper epitopes is usually robust, but also responses to the antigen of interest have been expanded.
Gerloni et al. demonstrated that linking a self-epitope (a Th-cell determinant of MUC.1) with NV epitope plasmid DNA expressing human antibody could create a form of associative recognition of antigen and enable the induction of an anti–self-epitope response based on help provided by the anti-NV epitope response (Langlade-Demoyen et al., 2003; Rizzi and Castiglioni, 2004). Further, different temporal dynamics of the two responses was found: the response against the NV epitope preceded the response against the self-epitope by approximately 48 h (Gerloni et al., 2000). The reasons of this difference in the kinetics of the response may be difference in (a) the affinity of the two peptides for MHC class II molecules, (b) the precursor frequency for each of the two determinants (this was determined to be low in both instances), or (c) an incremental activation of the APC (e.g., increased antigen processing/presentation) (Zanetti, 2005).
In the present study, we demonstrated that NV peptide, a strongly immunogenic MHC-II–restricted epitope, could augment the efficacy of gp100 peptide–pulsed DC immunization in mediating both successful immune priming toward and therapeutic rejection of B16 melanoma and its subline D5. Further, we found that both the cytotoxicity and the frequency of gp100-specific CTLs were increased after immunization with DCs(NV)-gp100. The NV peptide increases T-helper activation by augmenting IFN-γ and CD40L expression, which in turn increases activation of DCs and alters the DCs qualitatively, making them more Th1 polarizing through increased IL-12 production. The conditioned DCs, through higher IL-12, B7-1, and B7-2, not only more effectively activate CTLs and induce better protection against melanoma, but also feed back on the helper T cells to polarize them more toward the Th1 phenotype.
The recruitment of CD4+ T cell help to CD8+ T cell responses requires interactions between CD40 and CD40 ligand and is thought to occur through antigen-presenting cell (APC) activation (Ridge et al., 1998; Kalinski et al., 2006; López-Bravo and Ardavín, 2008). The generation of memory CD8+ T cells displaying an enhanced capacity for cell division and cytokine secretion also required CD4 help and CD40 expression by the APCs (Shedlock and Shen, 2003; Sun and Beven, 2003; Smith et al., 2004). CD8+ T cells receive CD4 help directly through CD40 and this interaction is fundamental for CD8+ T-cell memory generation (Shedlock and Shen, 2003; Sun and Beven, 2003). Our finding that MHC-II–restricted NV peptide could upregulate CD40 expression on DCs more potently than controls provides a reasonable explanation for the capacity of NV peptide to augment the immune response of gp100 peptide–pulsed DC vaccine. This finding is also consistent with a study showing that protective antitumor immunity required CD40-mediated activation of DCs (Schoenberger, 1998). Further, we also found more IFN-γ–producing CD8-positive T cells from the DC(NV)-gp100 group than from the DC(NV) and DC(gp100) groups. We believe that the gp100 peptide and NV peptide act synergistically with each other. Apart from that NV peptide could increase the number of gp100-specific CD8+ T cells through activating DCs, the gp100 peptide also enhanced the immune response to NV peptide and produced more NV peptide–specific CD4+ T cells. Also, we found that treatment with DC-NV plus DC-gp100 showed no significant higher protective and therapeutic efficacy compared with DC-gp100 (data not shown). This result is consistent with a previous study (Schoenberger, 1998) that demonstrated that Th-cell CTL priming must be provided in a cognate manner, such that both Th cells and the CTLs recognize antigen on the same APC. Second, combination of gp100 and NV peptide may ameliorate the immune microenvironment, reduce the threshold of immune response, and lead to increased cytokine secretion.
Thus, a key effect of enhanced Th epitope affinity is increased IL-12 production. IL-12 acts in driving the selective expansion of Th1 cells (Macatonia et al., 1995; Bennett et al., 1998; Diveu et al., 2008), enhances CTL proliferation (Trinchieri, 1998), and may further upregulate costimulation (Li et al., 2010). In our studies, we observed that the NV peptide clearly promotes the production of IFN-γ by T cells and skewed the Th phenotype to Th1. Thus, the IFN-γ secreted by T cells in turn upregulates IL-12 production by APCs and selection of a Th1 phenotype. In addition to CD40, DCs activated by CD4+ help and stimulated with NV peptide induced significantly higher levels of expression of other costimulatory molecules, such as B7-1 and B7-2, which also play vital roles in the induction and expansion of CTL's activity, indicating that the resulting increase in CTLs after immunization with DC(NV)-gp100 was related with upregulation of B7-1 and B7-2 by NV peptide.
Also, we did not find any autoimmune response or damage to the host, such as vertigo. We hypothesize that the reasons for this are (1) the expression of gp100 in melanoma is higher than the normal melanocytes and (2) the tumor-reactive CD8+ T cells elicited by the gp100 epitope used in our vaccine may be not shared with normal melanocytes. The specific mechanism will be further researched.
In summary, in this study we found that the NV peptide augments the efficacy of gp100 peptide–pulsed DC immunization. On exploring the mechanisms behind the epitope enhancement effect, we discovered several mechanisms by which NV peptide contributes to CTL induction and promotes the cytotoxicity of tumor antigen–specific CTL. Epitope enhancement to increase the affinity of a class II MHC binding peptide can dramatically enhance the efficacy of a Th-CTL peptide vaccine, constructed for CTL induction and protection against tumor challenge, both quantitatively and qualitatively. The NV peptide shifts the quality of the response toward Th1 by greater upregulation of IFN-γ by the helper T cells, which then more effectively condition and polarize the DCs by upregulation of B7-1 and B7-2 expression and IL-12 production, thereby improving CTL induction and skewing the cytokine profile. Better understanding of the mechanism of NV peptide may lead to the rational design of more effective tumor vaccines.
Footnotes
Acknowledgments
This work was supported by the National Key Technologies R&D Program of China during the 11th Five-Year Plan Period (2009ZX10004-104 and 2009ZX09301-011), National Basic Research Program of China (973 Program, 2010CB912600, 2011CB910400), National Science Foundation of China (30872378, 81072408), and the Science and Technology Commission of Shanghai Municipality (10JC1401100) in China.
Disclosure Statement
No competing financial interests exist.