
Abstract
The inflammatory responses in Alzheimer's disease and prion diseases are dominated by microglia activation. Scavenger receptors have been recently related to the innate immune activation of microglia initiated by endogenous ligands. In this study, we investigated mRNA expression patterns of B class scavenger receptors CD36 and scavenger receptor B1 (SR-B1) in BV2 microglia upon exposure to amyloid fibril Aβ1–42 and PrP106–126, respectively. CD36 and SR-B1 showed similar mRNA expression patterns following each treatment. PrP106–126 induced a rapid increase of CD36 and SR-B1 mRNA levels in the treated microglia, whereas Aβ1–42 induced a delayed but persistent increase in the mRNA expression of CD36 and SR-B1. These results suggest a possible involvement of CD36 and SR-B1 in microglial interaction with amyloidogenic fragments of beta-amyloid and prion proteins.
Introduction
The abnormal assembly and deposition of amyloid fibrils in the brain characterizes a diverse group of neurodegenerative disorders that includes Alzheimer's disease (AD), Parkinson's disease, Huntington's disease, and prion diseases (Ross and Poirier, 2004), many involving the degeneration of neurons and microglial activation (Block et al., 2007). Prion diseases and AD are neurodegenerative disorders neuropathologically characterized by the presence of extracellular amyloid deposits, extensive neuronal loss, and gliosis. In AD, the amyloid deposits are composed of amyloid-beta (Aβ) peptides (40–42 amino acids), which result from the abnormal proteolytic cleavage of the amyloid precursor protein (Yankner and Lu, 2009), whereas in prion diseases, the amyloid deposits are constituted mainly of the scrapie isoform of the prion protein (PrPSc), a conformational variant of the cellular prion protein (PrPC) (Aguzzi and Heikenwalder, 2006).
The role of class B scavenger receptors, particularly CD36, in the amyloid-induced microglial activation has been extensively investigated in AD (El Khoury et al., 1996, 2003; Paresce et al., 1996; Husemann et al., 2001; Husemann and Silverstein, 2001; Coraci et al., 2002; Moore et al., 2002; Srivastava and Jain, 2002; Bamberger et al., 2003; Ricciarelli et al., 2004; Stewart et al., 2010; Thanopoulou et al., 2010). As part of an ongoing study of the role of scavenger receptors in amyloid-related disorders in the central nervous system, we examined mRNA expression patterns of B class scavenger receptors CD36 and SR-B1 in the mouse microglia cell line BV2 upon exposure to the synthetic peptide Aβ1–42 (the major components of amyloid plaques in AD) or PrP106–126 (a peptide fragment that mimics PrPSc toxicity and forms fibrils in vitro), respectively. We found that prion peptides induced a rapid but transient increase of CD36 and SR-B1 mRNA levels in the treated microglia, whereas Aβ induced a delayed but persistent increase in the mRNA level of CD36 and SR-B1.
Materials and Methods
Beta-amyloid and prion protein peptide
The human sequence of the prion protein fragment (KTNMKHMAGAAAAGAVVGGLG; PrP106–126), the scrambled sequence (MEVGWYRSPFSRVVHLYRNGK; ScrPrP), Aβ1–42, and Aβ42–1 (a reverse form of Aβ1–42 used as negative control) were synthesized by Shanghai Sangon (China). Lyophilized prion peptides were dissolved in PBS (pH 7.4) at a concentration of 5 mM and stored at −20°C as stock solutions. Aβ1–42 or Aβ42–1 peptides were aged in PBS buffer, in a stock concentration of 100 μM, for 7 days at 37°C to obtain fibrillar Aβ.
Cell line and culture
BV2 cells, a murine microglia cell line, were maintained in a 95% air and 5% CO2 atmosphere in DMEM-F12 supplemented with 10% fetal bovine serum (FBS; Gibco), 100 μg/mL streptomycin, 100 U/mL penicillin (Gibco), and 2 mM glutamine.
Peptide treatment
Cultured microglial cells were treated with Aβ1–42 or Aβ42–1 (5 μM), PrP106–126 (50 μM), or scrambled PrP (50 μM) for time intervals ranging from 3 to 30 h, as indicated. The peptides were added into culture medium on the second culturing day.
RNA extraction and reverse transcription
The total RNA of treated cells was extracted using the TRIzol Reagent (Invitrogen) and treated with RNase-free DNase I to remove possible contaminating DNA. The purity of the total RNA was estimated by the OD260/OD280 absorbance ratio (1.9–2.0). Measurement of RNA concentration was conducted at OD260 nm on a spectrophotometer (BioPhotometer). Constant amounts of 1000 ng of RNA from each sample were subjected to reverse transcription (RT) to cDNA using the Reverse Transcription System (Promega). Six randomly chosen control samples without transcriptase (RT-negative) were used as controls for the RT step.
Real-time quantitative RT–polymerase chain reaction analysis of the expression of CD36 and SR-B1
To determine the mRNA expression of the receptors, real-time polymerase chain reaction (PCR) was carried out. Endogenous housekeeping gene β-actin was used as cDNA template control. PCR primers were used for amplification of genes cloned for real-time PCR (Table 1 and Fig. 1).
Electrophoresis of reverse-transcription polymerase chain reaction–amplified CD36 and SR-B1 on 8% agarose gel, which was then stained with ethidium bromide. A: β-actin, 223 bp; B: SR-B1, 257 bp; C: CD36, 294 bp. SR-B1, scavenger receptor B1.
For construction of a standard curve, a 294-bp fragment of the CD36 gene and a 257-bp fragment of the SR-B1 gene were cloned into pGEM-T-Vector by T-A cloning, and five 10-fold serial dilutions of plasmid DNA ranging from 102 to 106 molecules were then prepared.
Real-time quantitative PCR was performed using a DNA Engine Opticon™ 2 system (MJ Research) and DyNAmo™ SYBR Green quantitative PCR kit (MJ Research). The primers were used to amplify cDNA of the biological sample, negative controls, and five plasmid DNA standards. The PCR was carried out in a real-time PCR tube. The total volume was 20 μL, which includes 8.75 μL water, 0.3 μL of each primer (10 pmol), 10 μL master mix, and 25 ng of reverse-transcribed total RNA or plasmid DNA of the respective concentration. The PCR amplification was carried out as follows: after denaturation at 94°C for 5 min, 36 PCR cycles were performed, including 94°C for 30 s, 62°C for 20 s, 72°C for 20 s, and 1 s at 84°C appended for a single fluorescence measurement above the melting temperature of possible primer-dimers. Finally, a melting step was performed, consisting 10 s at 65°C and slow heating with a rate of 0.1°C per second up to 95°C, with continuous fluorescence measurement.
The mRNA level of CD36 and SR-B1 was calculated using an absolute standard curve method (Morrison et al., 1998; Pfaffl, 2001). The cDNA samples were amplified in parallel with plasmid standards in each run and their Ct values were plotted together with the standard curves, from which the normalized mRNA copy numbers were determined. All samples were analyzed in triplicate.
Statistical analysis of quantitative PCR
Comparison of treatment effects was carried out using one-way analysis of variance techniques with post hoc tests according to Tukey (SPSS 13.0). Data are expressed as means ± SD. Differences with p < 0.05 were considered statistically significant.
Results
RT and amplification of the target genes
RNA aliquots extracted from treated BV2 cells were reverse transcribed as described earlier, and the PCR was used to amplify cDNA specific for β-actin, CD36, and SR-B1 (Fig. 1).
The effect of prion peptides on mRNA expression of CD36 and SR-B1
BV2 cells were incubated with 50 μM of neurotoxic PrP106–126 or Scr PrP for 3, 6, 12, 24, and 30 h. The expression level of CD36 and SR-B1 mRNA was measured by real-time PCR. The results showed that CD36 and SR-B1 had identical expression patterns upon exposure to PrP106–126 (Fig. 2). No significant change in CD36 and SR-B1 mRNA levels was observed in BV2 cells treated with scrambled PrP or PBS (Fig. 2).
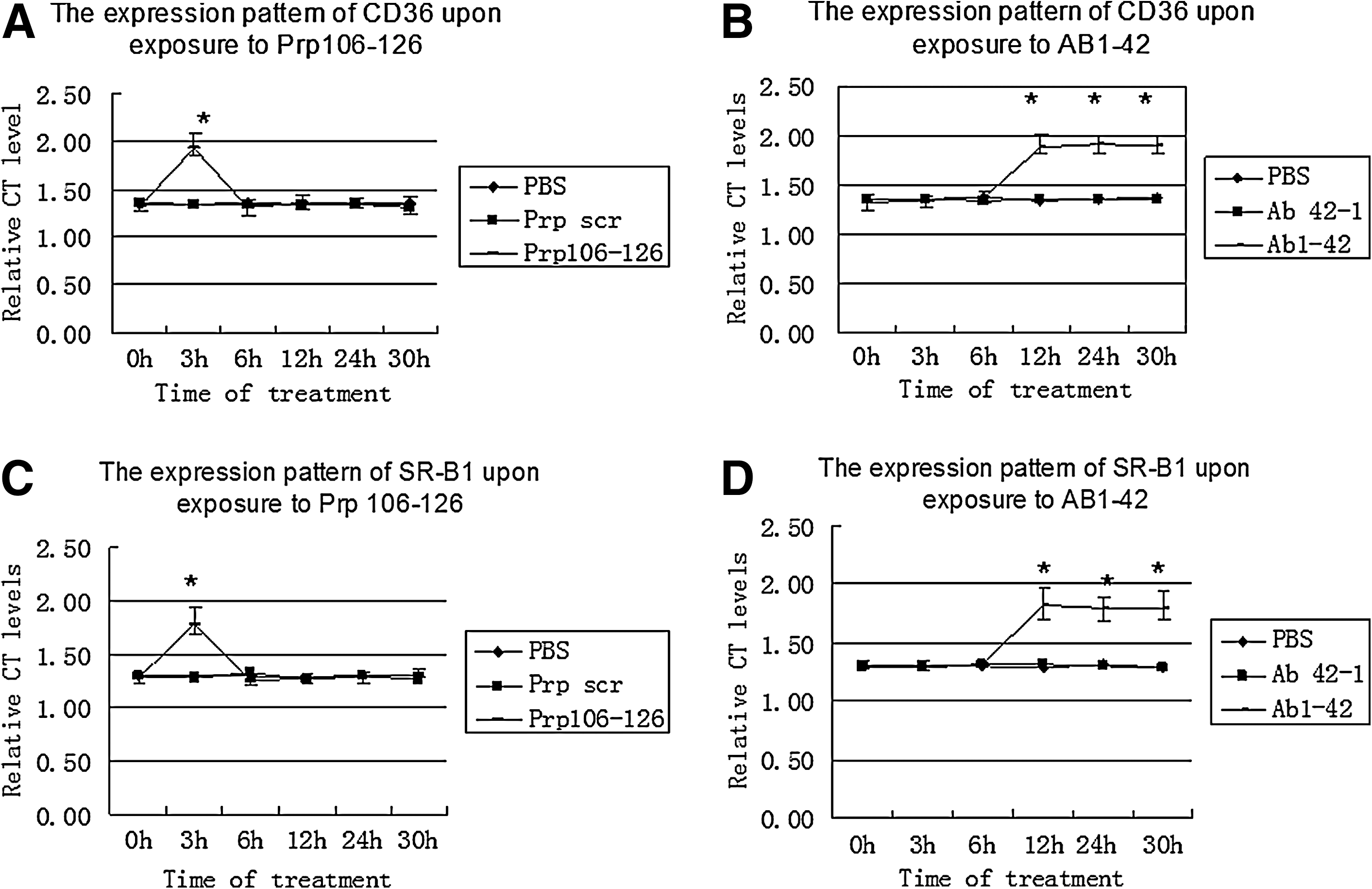
Messenger RNA expression of CD36 and SR-B1 after treatment with PrP106–126
The mRNA levels of CD36 and SR-B1 significantly increased at 3 h in the cells treated with PrP106–126 (p < 0.05) and were significantly higher than those in the cells treated with the scrambled PrP or PBS (p < 0.05). After 3 h, the mRNA levels of the two genes in PrP106–126-treated cells returned to and remained at baseline levels for the remainder of the experiment and showed no significant difference with mRNA levels in BV2 cells treated with the scrambled PrP or PBS (Fig. 2).
The effect of beta-amyloid on mRNA expression of CD36 and SR-B1
Analysis of CD36 and SR-B1 mRNA expression in BV2 after exposure to 5 μM of Aβ1–42 for different treatment times (3, 6, 12, 24, and 30 h) also revealed identical expression patterns of CD36 and SR-B1.No significant change in CD36 and SR-B1 mRNA levels was observed in BV2 cells treated with Aβ42–1 or PBS.
The mRNA levels of the two genes were significantly increased by 12 h of Aβ treatment (p < 0.05) and were then maintained at that high level for the remainder of the experiment, during which time they were significantly higher than the mRNA levels in the cells treated with Aβ42–1 or PBS (p < 0.05).There was no significant difference in CD36 and SR-B1 mRNA levels between BV2 cells treated with Aβ, Aβ42–1, or PBS, respectively, at 3 and 6 h (Fig. 2).
Discussion
The activation of microglia at sites of Aβ or PrPSc deposition is believed to result in a local, chronic inflammation that underlies the pathophysiology of AD and prion diseases (Combs et al., 1999). Aβ deposition induces scavenger receptors CD36 and SRB1 overexpression in microglia, which is directly linked to the oxidative stress associated with AD pathogenesis (Husemann et al., 2001; Ricciarelli et al., 2004). As the first part of an ongoing study of the role of scavenger receptors during sterile inflammation in the central nervous system, we compared the mRNA expression pattern of the genes of class B scavenger receptors in BV2 microglia after exposure to PrP106–126 and Aβ1–42. The results presented here show that CD36 and SR-B1 are expressed in microglia even without any stimulation. Although the mRNA expression level cannot be automatically associated with higher or lower protein expression, the observed results are consistent with many reports of constitutive expression of scavenger receptors in monocytes and macrophages (Hirano et al., 1999; Husemann and Silverstein, 2001; Coraci et al. 2002; Husemann et al., 2002).
The results also showed identical expression pattern of both CD36 and SR-B1 in the microglia treated with either PrP106–126 or Aβ1–42, respectively. In the case of PrP106–126, there is a significant but transient increase in mRNA levels of both receptors, whereas Aβ1–42 induced a delayed but persistent increase in the mRNA expression of CD36 and SR-B1. The observed identical expression patterns of CD36 and SR-B1 suggest that there may be a functional association between these two receptors. We speculate that SR-B1 may act as a coreceptor to CD36 or act as a regulator of CD36-induced signaling. The nature of this relationship deserves further investigation, particularly in the context of the diseases in which the two receptors are known to be involved, such as AD and atherosclerosis.
CD36 is involved in the pathogenesis of many endogenous ligand-induced inflammations such as atherosclerosis and AD (Varban et al., 1998; Husemann et al., 2002; Van Eck et al., 2007; Stewart et al., 2010). In the setting of AD, CD36 is involved as a component of a cell surface receptor complex that mediates the binding of microglia to Aβ fibrils and the subsequent activation of intracellular signaling. Interestingly, the principal components of this receptor complex are shared with those for other fibrillar proteins and thus represent general elements through which myeloid lineage cells recognize complex fibrillar proteins (Bamberger et al., 2003). The tendency of CD36 to act as a coreceptor was confirmed by recent reports that revealed its interaction with Toll-like receptors to form a complex receptor that promotes inflammation in atherosclerosis, AD, and fungal diseases (Means et al., 2009; Stewart et al., 2010). The observed persistent high level of CD36 mRNA upon exposure to Aβ1–42 is consistent with the findings observed in the aforementioned reports. The significant, albeit transient, increase in CD36 mRNA expression following exposure to fibrillar prion peptide leads us to speculate that CD36 may act also as a receptor, most probably as a component of the complex receptor that mediates PrP106–126-induced microglial activation. This is supported by recent findings from our laboratory, which demonstrate that the mAb blockade of CD36 abrogates PrP106–126-induced IL-1β and iNOS upregulation in BV2 microglial cells, but has no effect on PrP106–126-induced nuclear factor kappa B or capase-1 activation, suggesting that nuclear factor kappa B activation may not be a key element in the signaling pathways through which CD36 mediates upregulation of pro-inflammatory molecules in PrP106–126-treated BV2 microglia (Kouadir et al., 2011, in review).
SR-B1 has also been implicated in the pathogenesis of atherosclerosis, AD, and viral diseases such as hepatitis C virus (Areschoug and Gordon, 2009). The observed increase of mRNA level of SR-B1 in microglial cell after exposure to Aβ1–42 confirms previous reports of its involvement in beta-amyloid–induced microglial activation. The significant, albeit transient, increase in SR-B1 mRNA expression following exposure to PrP106–126 suggests a possible role of this receptor in PrP106–126-induced microglial activation. Finally, the differential expression pattern of CD36 and SR-B1 upon exposure to PrP106–126 or Aβ1–42 suggests that these two receptors may be implicated, with different kinetics, in Aβ- and prion peptide-induced microglial activation.
Footnotes
Disclosure Statement
No competing financial interests exist.