
Abstract
Bifunctional small hairpin RNAs (bi-shRNAs) are functional miRNA/siRNA composites that are optimized for posttranscriptional gene silencing through concurrent mRNA cleavage-dependent and -independent mechanisms (Rao et al., 2010). We have generated a novel bi-shRNA using the miR30 scaffold that is highly effective for knockdown of human stathmin (STMN1) mRNA. STMN1 overexpression well documented in human solid cancers correlates with their poor prognosis. Transfection with the bi-shSTMN1–encoding expression plasmid (pbi-shSTMN1) markedly reduced CCL-247 human colorectal cancer and SK-Mel-28 melanoma cell growth in vitro (Rao et al., 2010). We now examine in vivo the antitumor efficacy of this RNA interference-based approach with human tumor xenografted athymic mice. A single intratumoral (IT) injection of pbi-shSTMN1 (8 μg) reduced CCL-247 tumor xenograft growth by 44% at 7 days when delivered as a 1,2-dioleoyl-3-trimethyl-ammoniopropane:cholesterol liposomal complex. Extended growth reductions (57% at day 15; p < 0.05) were achieved with three daily treatments of the same construct. STMN1 protein reduction was confirmed by immunoblot analysis. IT treatments with pbi-shSTMN1 similarly inhibited the growth of tumorgrafts derived from low-passage primary melanoma (≥70% reduction for 2 weeks) and abrogated osteosarcoma tumorgraft growth, with the mature bi-shRNA effector molecule detectable for up to 16 days after last injection. Antitumor efficacy was evident for up to 25 days posttreatment in the melanoma tumorgraft model. The maximum tolerated dose by IT injection of >92 μg (Human equivalent dose [HED] of >0.3 mg/kg) in CCL-247 tumor xenograft-bearing athymic mice was ∼10-fold higher than the extrapolated IC50 of 9 μg (HED of 0.03 mg/kg). Healthy, immunocompetent rats were used as biorelevant models for systemic safety assessments. The observed maximum tolerated dose of <100 μg for intravenously injected pbi-shSTMN1 (mouse equivalent of <26.5 μg; HED of <0.09 mg/kg) confirmed systemic safety of the therapeutic dose, hence supporting early-phase assessments of clinical safety and preliminary efficacy.
Introduction
The effectiveness of STMN1-targeted approaches has been mostly examined in vitro by us (Rao et al., 2010) and others (Zhang et al., 2006; Wang et al., 2007; Jiang et al., 2009; Carney and Cassimeris, 2010; Gan et al., 2010; Hsieh et al., 2010; Jeon et al., 2010) as singlet or combinational therapy with chemotherapeutic agents (Iancu et al., 2000; Alli et al., 2002; Mistry and Atweh, 2006; Mistry et al., 2007; Wang et al., 2007; Alli et al., 2007a; Rayburn and Zhang, 2008; Vaishnaw et al., 2010). On the basis of cumulative findings that STMN1 knockdown correspondingly inhibited human tumor cell growth, we have constructed a novel RNA interference (RNAi)–based therapeutic with bifunctional features. The bifunctional small hairpin RNA (bi-shRNA) comprises of two stem-loop structures encoded within the same expression construct. The transcribed mature effector small RNA molecules are endowed with separate posttranscriptional cleavage-dependent or -independent gene silencing functions. Using this bifunctional shRNA specific for STMN1 (pbi-shSTMN1), we have demonstrated effective STMN1 knockdown in human colorectal cancer CCL-247 cells, achieving significant tumor cell kill at enhanced potency (IC50 that was 5 log lower than molar equivalent of RNA oligonucleotides) and for a longer duration than previously described olignonucleotide-based approaches. Treated CCL-247 cells with a wild-type p53 phenotype (Liu and Bodmer, 2006; Olivier et al., 2009) displayed cell-cycle arrest and subsequent apoptosis (Rao et al., 2010). Increased STMN1 expression was associated with p53 mutation that contributed to enhanced tumorigenicity in hepatocellular carcinoma (Yuan et al., 2006).
The present study assessed the in vivo efficacy of intratumoral (IT) pbi-shSTMN1 treatments, using immune-compromised mice bearing xenografts derived from established human cancer cell lines or low-passage primary human tumors. Additionally, safety of the pbi-shSTMN1-LP was assessed following a single intravenous (IV) injection in a biorelevant rat model. With an ultimate aim of achieving systemic cancer-selective delivery, studies were performed following encapsulation of the pbi-shSTMN1 in cationic liposomes composed of the biodegradable lipid 1,2-dioleoyl-3-trimethyl-ammoniopropane (DOTAP) and cholesterol (pbi-shSTMN1-LP). Cationic liposomes are one of the most commonly used nonviral vehicles for cancer therapeutics delivery (Templeton, 2002; Zhang et al., 2007; Ashihara et al., 2009) and known to prolong the retention time of plasmid DNA in tumors when compared with administration of naked DNA (Clark et al., 2000; Nomura et al., 1997). pbi-shSTMN1 plasmid was encapsulated in DOTAP:cholesterol liposomal formulation at a ratio of 50:45, previously shown to be highly effective for achieving gene expression of the plasmid DNA payload administered intramuscularly (Phadke et al., 2009), intratumorally (Ramesh et al., 2001; Templeton, 2002) or intravenously (Templeton et al., 1997).
Materials and Methods
Plasmid DNA and preparation of lipoplexes
The pbi-shSTMN1 plasmid and the scrambled control plasmids were constructed as previously described (Phadke et al., 2009; Rao et al., 2010). An equal volume of plasmid DNA (1 mg/mL) was mixed with 2X DOTAP:cholesterol to form the final plasmid DNA-Lipoplex suspended in D5W (diluent consisting of 5% dextrose in water). Empty liposomes were prepared by diluting stock DOTAP:cholesterol in D5W to a final concentration of 1X. Prepared lipoplexes were stored in sterile vials at +2°C–8°C in a dark container, until ready for use. Lipoplexes were tested for sterility, endotoxin, and optical characteristics (optical density 400, particle size, and zeta potential) before final release.
Antitumor efficacy
In one study, 5- to 6-week-old female athymic nude (
Determinations on low-passage tumorgrafts derived from patient tumor were performed with athymic nude mice bred from an internal breeding colony (established from mice purchased from Charles River). The animals were housed in an animal facility approved by IACUC at Van Andel Research Institute (VARI). The tumor models in the Human Tumor Xenograft Bank at VARI were established by grafting tumor biopsy specimens from patients diagnosed with cancer into immune-compromised mice. These tumors are henceforth referred to as “tumorgrafts” to differentiate them from “tumor xenografts” established using tumor cell lines grown in vitro. The resulting tumorgrafts were subsequently cryopreserved and profiled molecularly on the Human Genome U133 Plus 2.0 Array (Affymetrix). Genomic analyses were performed using Xenobase-BioIntegration Solutions, a bioinformatics package developed at VARI to manage and analyze data across molecular, cellular, preclinical, and clinical platforms (
Mice were intratumorally injected when the tumors reached an approximate size of 100 mm3 (CCL-247 tumor xenografts), 260 mm3 (osteosarcoma tumorgrafts), and 160 mm3 (melanoma tumorgrafts). IT injections were performed either singly or at 3 or 6 consecutive days or semiweekly for 3 weeks. pbi-shSTMN1-LP was administered at doses ranging from 0.007 to 92 μg (depending on the study) in total volumes ranging from 100 to 200 μL. Controls included lipoplex containing a scrambled (sc) control, empty liposomes without plasmid DNA, and diluent (D5W: water + 5% dextrose) administered in equivalent volumes. The tumors (either xenograft or tumorgrafts) were measured using Vernier calipers postinjection when they became palpable. Tumor volumes were calculated with the following formula: (L × W 2) × 0.5, where L is length and W is width of the tumor. All mice were monitored and tumors were measured on alternate days. Tumors were harvested and weighed on the day of sacrifice. A major part of each tissue was snap frozen in dry ice (for molecular analysis) and a small tissue piece immersed in 10% buffered formalin. The formalin-fixed tissue was paraffin embedded and hematoxylin and eosin (H&E)–stained slides were prepared. Histopathology analysis was performed by Propath Labs (mouse tumor tissues) and by IDEXX laboratories (mouse organ tissues). Assessments were based on comparisons of area distribution of viable versus necrotic areas of H&E-stained specimens by standard histopathologic criteria.
In the melanoma tumorgraft model, tumor growth progression to a surrogate endpoint was analyzed using a Kaplan–Meier estimator (Rouleau et al., 2008). The surrogate endpoint was a melanoma tumorgraft size of 420 mm3 (a size that was 50% of the average tumor size for the D5W group at the time of sacrifice).
Toxicology analysis
Six- to 7-week-old male and female Sprague–Dawley rats (Harlan Laboratories) were housed in an animal facility approved by the IACUC at UNT, Health Science Center. The animals were divided into five treatment cohorts of 60 rats (30 male and 30 female) each. Three groups were given pbi-shSTMN1-LP at one of three doses—1, 10, and 100 μg in a total volume of 300 μL. Two groups served as controls and were injected with either empty liposomes or D5W. A 5 mM DOTAP:cholesterol formulation in D5W was used for DNA encapsulation or tested as empty-liposome control at the equivalent test volume used to encapsulate 100 μg of pbi-shSTMN1. Rat body weight was measured once every week for the duration of the study. Ten rats (five male and five female) from each group were sacrificed at one of six time-points (days 2, 7, 14, 30, 60, or 90) posttreatment, and blood and internal tissues were harvested. Blood was collected for toxicology analysis: complete blood count (white blood cells [total and differential], red blood cell count, hemoglobin concentration, hematocrit, platelet counts, mean corpuscular volume, mean corpuscular hemoglobin concentration, and mean corpuscular hemoglobin), serum chemistry (alanine aminotransferase [ALT], aspartate aminotransferase [AST], creatinine, blood urea nitrogen, bilirubin, creatinine kinase, albumin, electrolytes, alkaline phosphatase, and glucose), and coagulation tests. Formalin-fixed, paraffin-embedded tissues were sectioned and H&E stained for histopathology analysis (Animal Resource Center at UT Southwestern Medical Center). Blood work analysis was performed by Comparative Clinical Pathology Lab at Research Animal Diagnostic Laboratory. Histopathology analysis of rat tissues was performed by the Cardiovascular Pathology Laboratory, Texas A&M University.
Immunoblotting
Immunoblotting was performed on frozen CCL-247 tumor xenografts treated with either pbi-shSTMN1-LP or D5W as outlined earlier (Rao et al., 2010). Briefly, for each particular experiment, lysates were diluted to the same protein concentration (25 μg protein per well) and denatured via addition of β-mercaptoethanol–containing Laemmli sample buffer (Bio-Rad) and incubation at 100°C for 5 min. Western blot was performed using 10% Tris-HCl gel (Bio-Rad). The primary antibody was a mouse monoclonal antibody to human STMN1 (Opt18 E3) (sc-55531, 1:1000) or β-actin (sc-47778, 1:100,000) (Santa Cruz Biotech). The secondary antibody was horseradish peroxidase–conjugated goat antibody to mouse IgG (sc-2005, 1:2500; Santa Cruz Biotech). Development was done using the chemiluminescence detection kit (SuperSignal® West Dura Extended Duration Substrate; Thermo Scientific). Band quantification was done using G:Box software (Syngene). STMN1 protein values were normalized relative to β-actin values for each sample. Total STMN1 knockdown for each xenograft sample was determined relative to untreated CCL-247 cells cultured in vitro.
Detection of mature shRNA by stem-loop RT-qPCR
Frozen osteosarcoma tumorgraft samples (∼30–60 mg) were homogenized for extraction of total RNA (including miRNA and shRNA) using mirVana (Ambion). Isolated total RNA was quantified using the NanoDrop spectrophotometer (Thermo Scientific). Three micrograms of total RNA was reverse transcribed in 40 μL final volume to produce cDNA using SuperScriptIII kit (Invitrogen) and guide strand–specific stem-loop reverse transcription (RT) primer (5′ GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAGGCA 3′). One microliter of synthesized cDNA (from 75 ng of total RNA) was used as template in the quantitative polymerase chain reaction (qPCR) with the primer pairs 5′ GCCCTTTGGCAGCCATTTG 3′ and 5′ GTGCAGGGTCCGAGGT 3′. The quantity of qPCR amplicon was measured using SYBR green fluorescent dye (Bio-Rad). A serial dilution of plasmid C10-421 (a plasmid construct containing the final 59 bp amplicon) was used in the qPCR to create the standard curve. The assay sensitivity was established at 100 copies of mature shRNA to exclude false-positive results that could result from nonspecific amplification.
Statistical analysis
All posttreatment tumor measurements were normalized to pretreatment values for individual tumors to enhance intergroup comparative analyses. Two-tailed Student's t-test analyses were used to compare percentage (%) of tumor growth reduction and tumor weights at necropsy between pbi-shSTMN1–treated group and D5W-treated cohort. All data analyses for the toxicology study were performed using SPSS 13.0 software (SPSS, Inc.). Differences in rat body weights and various complete blood count and serum chemistry parameters were analyzed at specific time points (days 2, 7, 14, 30, 60, and 90) or within a dose group by one-way analysis of variance. Post hoc comparisons were made using the Tukey test to identify specific groups varying in these parameters. For histopathology analysis for toxicology study, all morphologic diagnoses were reduced to a grouped value for each target tissue (normal, minimal/mild, moderate, or severe); then statistical analyses were performed using Kruskal–Wallis analysis of variance (two-tailed, Chi-square analysis for nonparametric data). Time to tumor growth progression was analyzed using a Kaplan–Meier estimator (SPSS 13.0).
Results
CCL-247 tumor xenograft growth reduction by single IT treatment with pbi-shSTMN1-LP
The pbi-shSTMN1 plasmid comprises DNA encoding for STMN1-targeting bi-shRNAs that is inserted into the SalI and BglII sites of the pUMVC3 expression vector under the control of an enhanced (pol II) CMV promoter (Pizzorno et al., 1988) and has been extensively characterized with respect to its biochemical features (Rao et al., 2009). We observed >90% depletion of the targeted STMN1 protein in lipofected CCL-247 human colorectal cancer cells, with corresponding growth inhibition of >80% at 48 h. pbi-shSTMN1 plasmid was similarly effective in inhibiting the growth of human breast cancer (MDA-MB-231) and melanoma (SK-MEL-28) cells by 45% and 48%, respectively (data not shown). Both MDA-MB-231 and SK-MEL-28 are known to carry missense p53 mutations (Bartek et al., 1990; Girnita et al., 2000). Thus, the pbi-shSTMN1 vector was highly effective for inhibiting the growth of human cancer cells with wild-type or mutant p53.
Initial in vivo efficacy assessments were performed following a single IT injection of the lipoplexed pbi-shSTMN1 on previously established CCL-247 xenografts (Fig. 1a). We observed significant reductions of CCL-247 xenograft growth of 44% at day 7 and 55% at day 8 postinjection with 8 μg of pbi-shSTMN1-LP when compared with untreated tumors (p < 0.05, n = 5). Control expression plasmid lipoplex (scrambled LP) did not significantly alter xenograft growth (Fig. 1a). Thus, a single IT treatment of 8 μg of pbi-shSTMN1-LP was effective to achieve tumor reduction for >7 days.
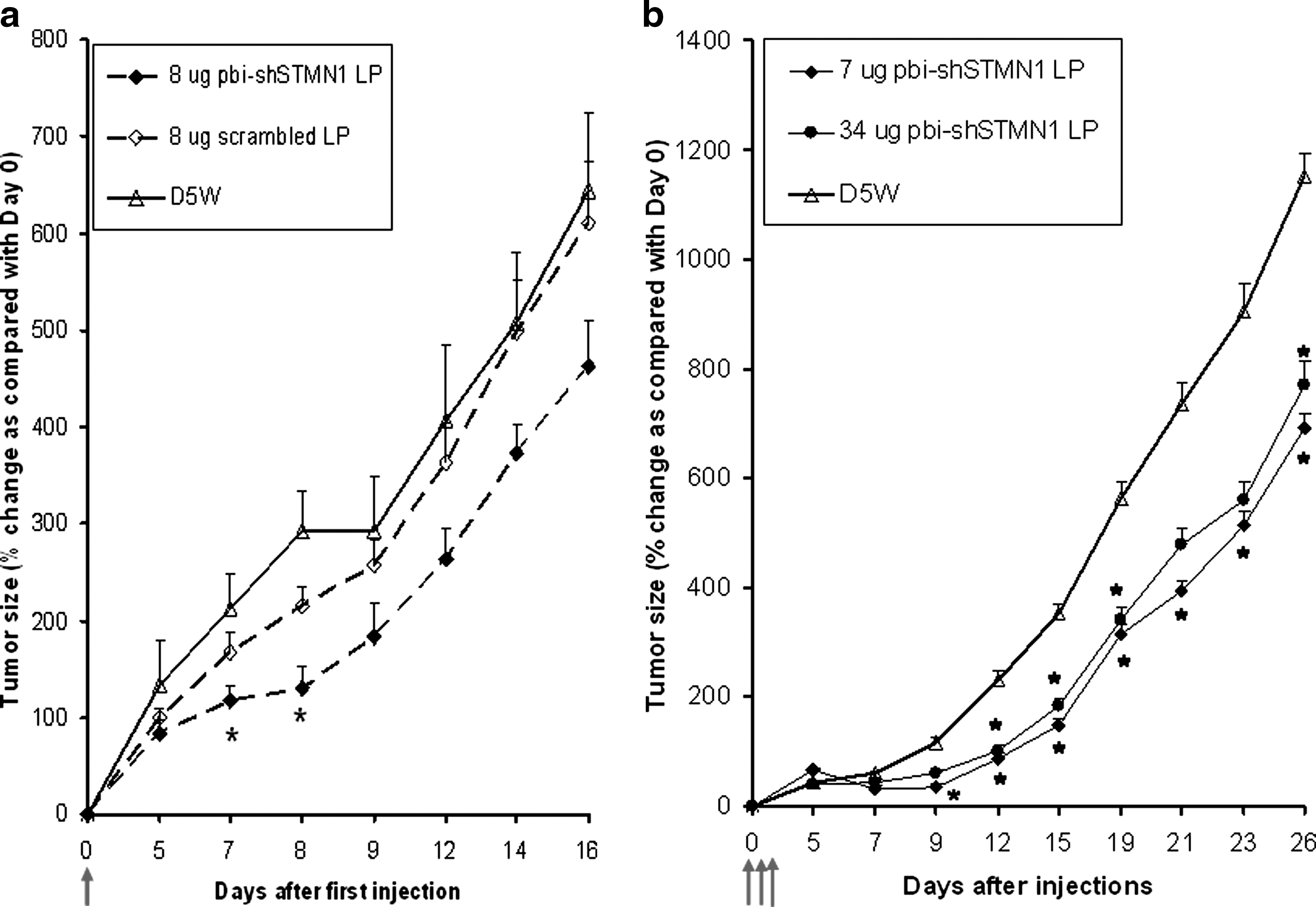
CCL-247 xenograft growth inhibitory activity following IT injection of pbi-shSTMN1-LP.
Prolonged growth inhibitory activity through repeat IT treatments with pbi-shSTMN1-LP in CCL-247 tumor xenografts
To optimize antitumor efficacy through repeat injections, CCL-247–xenografted mice were treated with three daily doses of pbi-shSTMN1-LP after tumors reached a size of ≥100 mm3. Compared with the earlier study wherein animals received only a single injection of the same dose, significant growth reductions were extended to 26 days after first injection in the 7 μg treatment arm (40% reduction; p < 0.05, n = 8), indicating that repeat treatment likely prolonged antitumor activity (Fig. 1b). Although dose-dependent outcomes were observed from 0.007 to 7 μg (0%–57% reductions at day 15 after first injection; R 2 = 0.86, linear regression analysis) (Fig. 2), only 7 and 34 μg treatments consistently attained significant growth reductions (57% and 48% reduction at day 15, respectively; p < 0.05, n = 8). Treatments with 0.01 μg (p = 0.6), 0.07 μg (p = 0.34), and 0.7 μg (p = 0.07) did not significantly alter tumor growth. These findings were confirmed by tumor weight measurements on day 26 after the first injection, with significant reduction in tumor weight for animals treated with 34 μg (0.65 ± 0.17 g; p = 0.03) and 7 μg (0.67 ± 0.13 g; p = 0.04) pbi-shSTMN1-LP when compared with D5W controls (1.00 ± 0.07 g).

Dose-dependent inhibitory activity of pbi-shSTMN1-LP using CCL-247 tumor xenografts. CCL-247 tumor xenografts in athymic nude mice were injected once daily for 3 consecutive days. Values represent mean ± SEM on day 15. CCL-247 tumor xenografts were injected with 0.01, 0.07, 0.7, 7, and 34 μg pbi-shSTMN1-LP or with diluent (D5W) (n = 8 for each group). Diluent (D5W)-treated tumors attained 352% of its original size at day 15. *p ≤ 0.05.
In vivo knockdown of targeted STMN1
To confirm the in vivo molecular impact of pbi-shSTMN1-LP treatment, we examined STMN1 expression in harvested control and treated CCL-247 xenografts by immunoblot analysis. Cell lysates prepared from in vitro cultured CCL-247 cells were used as a reference control for the immunoblots. STMN1 expression in cell lysates from treated tumors was compared with protein concentration–matched samples of excised mock-treated tumors at 24 h after six consecutive IT injections (Fig. 3a, b). pbi-shSTMN1-LP–treated tumors displayed significantly reduced STMN1 expression (44%–49%; 44% median reduction when compared with untreated CCL-247 cells; 3/3 tumors tested), according to densitometric measurements normalized to β-actin expression. In contrast, D5W-treated cohorts treated with LPs with a scrambled bi-shRNA–encoding plasmid payload did not show any significantly depleted STMN1 in the representative immunoblot (Fig. 3a).
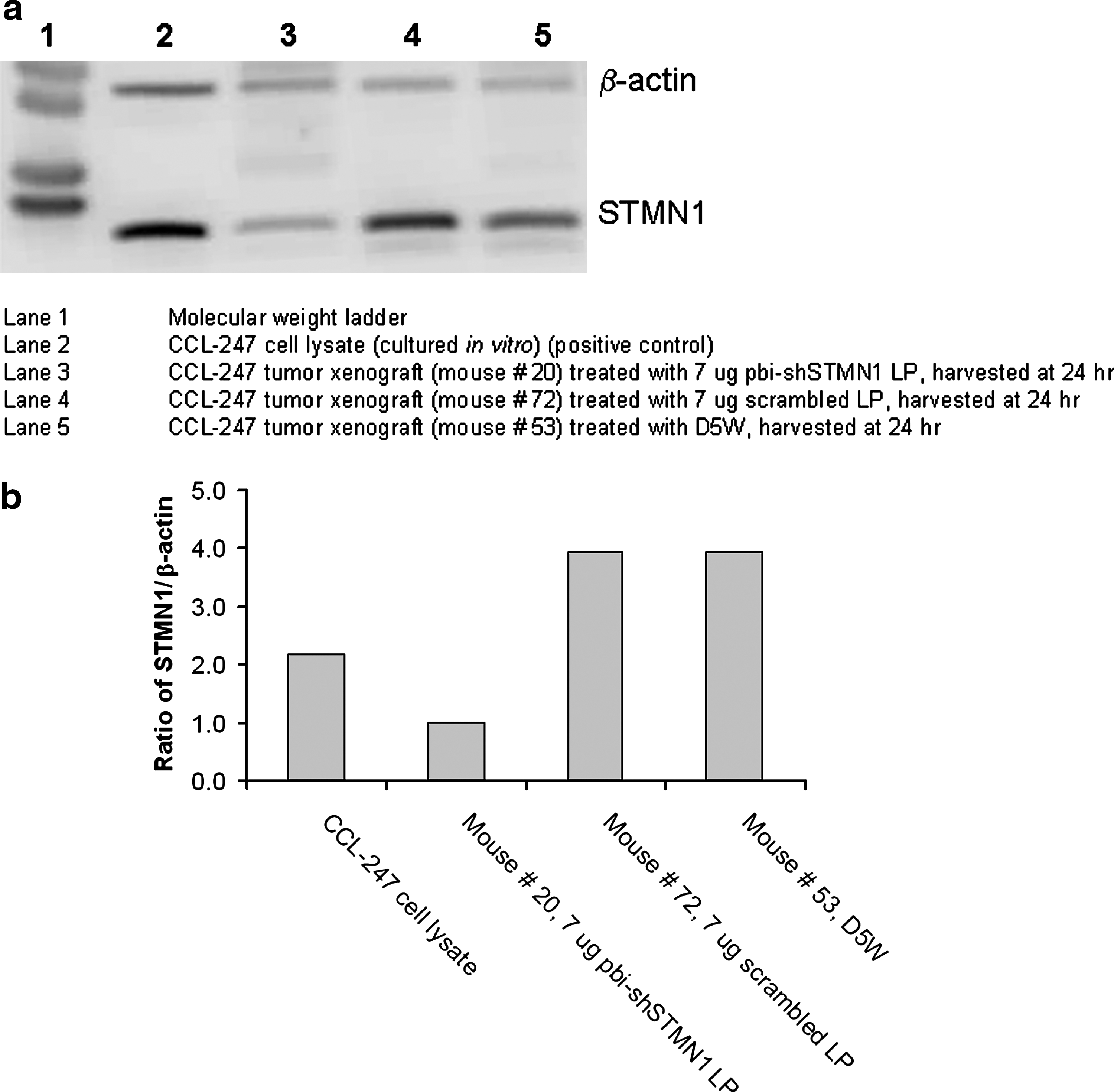
In vivo STMN1 expression following pbi-shSTMN1-LP treatment in CCL-247 tumor xenografts.
Antitumor efficacy of pbi-shSTMN1-LP in low-passage primary tumorgrafts
Recent studies showed that heterotransplanted primary human tumors can serve as clinically relevant models of investigation as they resemble the originating tumor in terms of pathology, tumor marker expression, interaction of tumor and stromal cells, and signal transduction pathways (Fu et al., 1992; Perez-Soler et al., 2000; Lopez-Barcons, 2009; Ding et al., 2010). Tumor models established from primary human pancreatic cancer, gastrointestinal stromal tumor, and glioblastoma multiforme were more correlative predictors with respect to clinical efficacy of novel cancer therapeutics (Fu et al., 1992; Lopez-Barcons, 2009; Revheim et al., 2009).
To this end, confirmatory in vivo antitumor efficacy studies on pbi-shSTMN1-LP were performed with low-passage primary tumorgrafts derived from surgically excised human melanoma and osteosarcoma tumors generated at VARI. Based on our findings of improved tumor efficacy of repeat injections in CCL-247 xenografts, six IT treatments of pbi-shSTMN1-LP were administered to the primary melanoma tumorgraft PTSH-0021 semiweekly for 3 weeks because of slow growth rate of the tumor (Fig. 4a). pbi-shSTMN1-LP (8 μg) significantly reduced tumor growth from day 20 (41%, p ≤ 0.05, n = 7) to 34 (53%, p < 0.05) after first injection. An increased dose of 40 μg attained stronger growth inhibition (70% reduction when compared with mock-treated tumors, p < 0.05 on day 26, n = 8) that was extended to day 46 (Fig. 4a). By comparison, similar treatments at 0.8 μg produced measurable tumor size reductions, which did not significantly differ from mock-treated tumors (33% at day 34 after first injection) or from treatments by empty liposome without an expression plasmid load (23% at day 34 after first injection). Tumor weight determinations confirmed that treatment with 40 μg pbi-shSTMN1-LP only significantly reduced tumor mass (0.46 ± 0.07 g; p = 0.02) when compared with D5W controls (0.85 ± 0.11 g) on the day of necropsy (day 46 after the first injection). Significantly reduced tumor mass was not observed after treatments with 0.8 or 8 μg pbi-shSTMN1-LP (0.56 ± 0.13 and 0.55 ± 0.06 g, respectively). The effect on tumor suppression at the 40 μg dose appeared most significant during the time of treatment (i.e., between days 0 and 20).
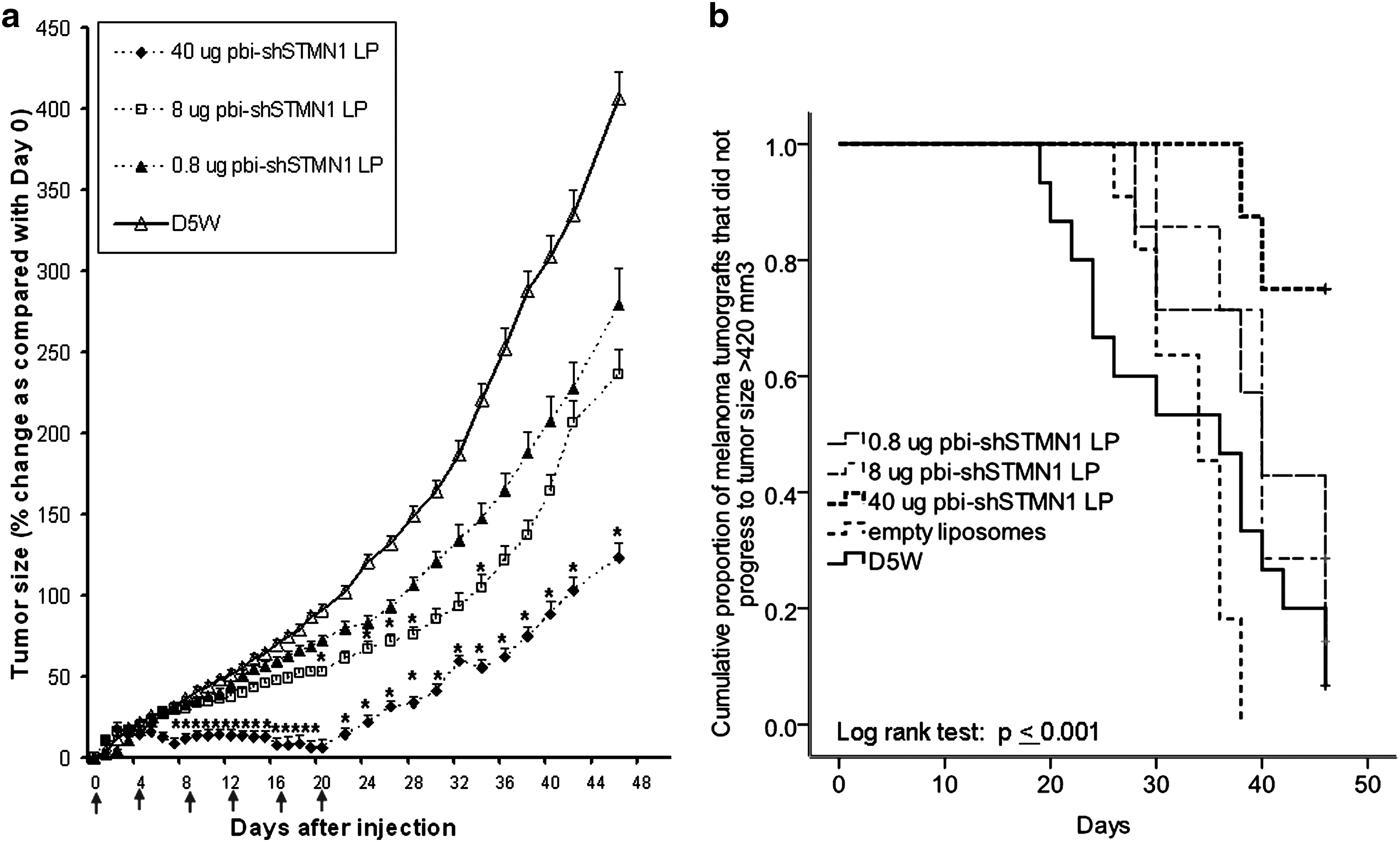
Antitumor activity of pbi-shSTMN1-LP against low-passage human tumorgrafts.

Kaplan–Meier analysis demonstrated that the median time taken by animals treated with control reagents to reach a tumor size of 420 mm3 (surrogate indicator) was 34 and 36 days (empty liposome and D5W groups, respectively), compared with 40 days for the animals treated with the lower doses (0.8 and 8 μg pbi-shSTMN1-LP). In contrast, the median time for the 40 μg group was not reached, as only 25% of the animals reached the surrogate endpoint, hence demonstrating a marked improvement over the controls (p < 0.001) (Fig. 4b).
In another study using osteosarcoma tumorgrafts, six injections of 46 and 92 μg of pbi-shSTMN1-LP essentially abrogated the growth (p < 0.05, n = 8) when compared with mock-treated tumorgrafts for up to 22 days after the first injection (16 days after last injection) (Fig. 4c). The last evaluable statistical time point was day 22 after the first injection in accordance with the maximum tumor size of the control cohort before termination of study, as outlined in the study protocol.
Extended bioactivity of pbi-shSTMN1
To correlate in vivo efficacy with intracellular expression of bi-shSTMN1, a time course determination was carried out to identify the presence of mature shRNA effector transcripts in treated osteosarcoma tumorgrafts (Fig. 5). Tumor tissues harvested from animals that were sacrificed from days 11, 20, and 22 after first injection (equivalent to 5, 14, and 16 days after last IT injection) all displayed mature, effector shRNAs, according to stem-loop RT-qPCR determinations with sequence-specific primer/probes (Fig. 5). Seven of eight treated tumors that were harvested between days 23 and 82 after first injection (corresponding to 17 and 76 days after last injection, respectively) did not contain detectable levels of shRNA. Thus, one of three animals treated with 92 μg of pbi-shSTMN1 demonstrated detectable copies of shRNA at day 54 after first injection (i.e., day 48 after last injection) (Fig. 5).
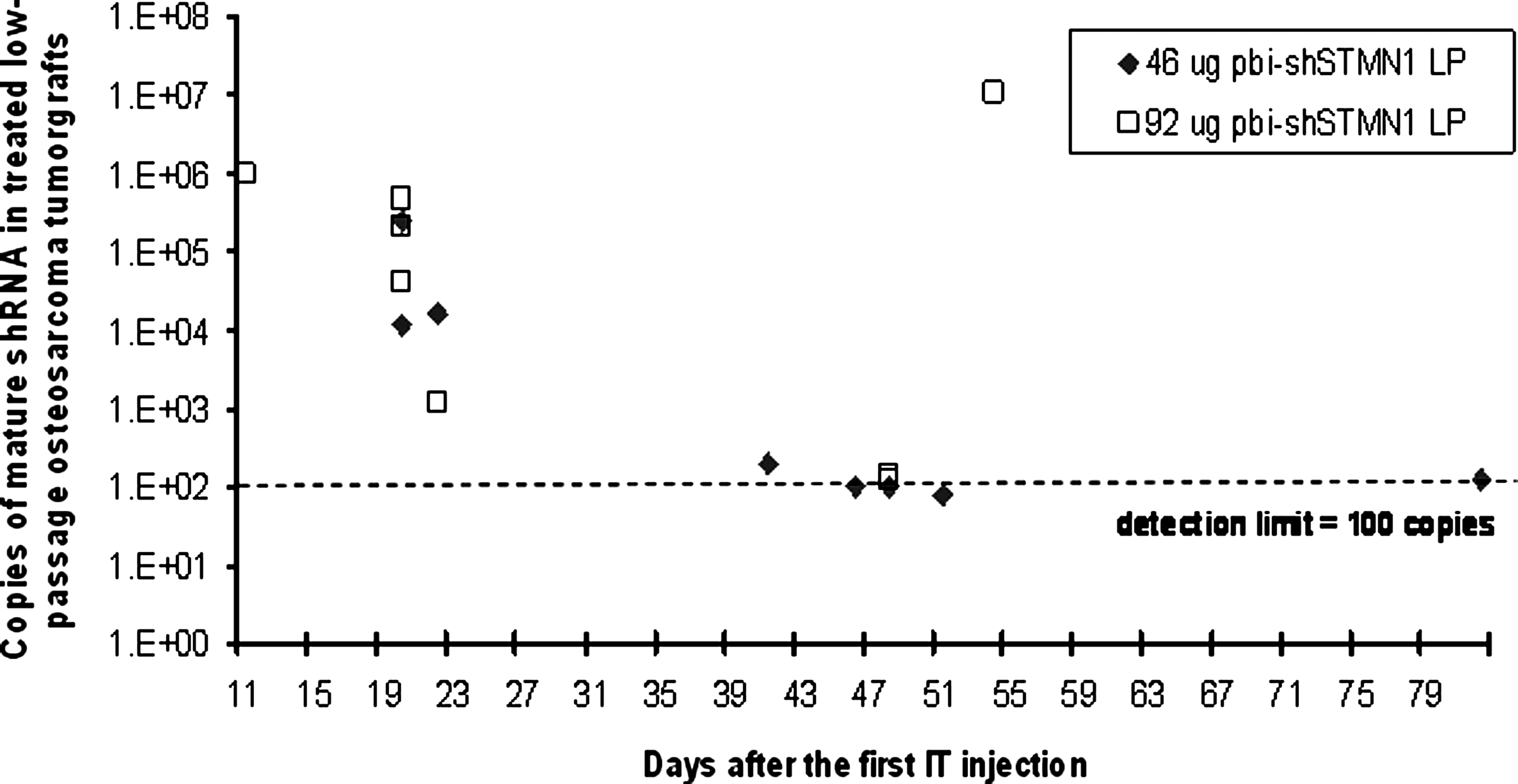
Mature shRNA expression in osteosarcoma tumorgrafts treated with 46 μg pbi-shSTMN1-LP (n = 8) or 92 μg pbi-shSTMN1-LP (n = 7). Each symbol corresponds to expression in a single animal. Detection limit of the assay was established at 100 copies. shRNA, small hairpin RNA.
As expected, tumors treated with scrambled LP, D5W, and empty liposomes did not display detectable levels of mature shRNA (data not shown). The extended expression of bi-shSTMN1 (up to day 16 after last injection) is an explanation for the prolonged antitumor activity of this treatment approach.
Histopathological assessments were performed on necropsied osteosarcoma tumorgrafts. Overall, residual tumorgrafts after pbi-shSTMN1-LP treatment (in either 46 or 92 μg cohorts) displayed reduced viability (mean value of 50%) when compared with tumorgrafts treated with the scrambled control (mean viability of 75%) and untreated tumors (mean viability of ∼95%). There were no distinguishing, treatment-related histopathological indications in any of the major mouse organ tissues examined (Table 1).
Systemic safety analysis in immunocompetent rats, a biorelevant model of human STMN1 knockdown
We have confirmed that rats are a biorelevant model for the purpose of toxicological determinations of pbi-shSTMN1-LP, based on observations that pbi-shSTMN1-LP knocked down rat STMN1 in rat cancer cell lines. In addition, a truncated rat sequence containing the bi-shSTMN1 target sequence was transiently transfected into CCL-247 cells and knocked down by pbi-shSTMN1-LP (data not shown). Cohorts of 60 healthy rats (30 males and females, each) received pbi-shSTMN1-LP (1.0, 10, or 100 μg; murine equivalent doses of 0.26, 2.6, and 26.5 μg, respectively) by a single IV injection and were sacrificed at graded time points (days 2, 7, 14, 30, 60, or 90). pbi-shSTMN1-LP was well tolerated at doses of 1.0 and 10 μg. However, 1 of 60 animals treated with pbi-shSTMN1-LP at a dose of 100 μg expired within 24 h of injection. Fifteen of the remaining 59 surviving rats demonstrated behavioral changes, which resolved by 24 h. None of the surviving rats demonstrated any toxicity beyond 24 h, and there were no histopathological changes in major organs at subsequent time points. As expected, treatment with the empty-liposome delivery vehicle also did not produce toxicity. Rat body weights were not significantly affected by any of the treatment doses of pbi-shSTMN1-LP.
Hematologic analyses confirmed an absence of acute or chronic toxicity by pbi-shSTMN1-LP. There was neither remarkable dose- nor time-dependent alterations for coagulation time, electrolyte levels, levels of glucose, or liver enzymes, or kidney function measurements (creatinine, urea nitrogen, and creatine kinase). We observed isolated elevations in creatine kinase levels at day 90 for the highest dose group (100 μg pbi-shSTMN1-LP, n = 4/10 vs. D5W, n = 0/10) and elevated AST levels for the lowest dose group (1 μg pbi-shSTMN1-LP, n = 10/10 vs. D5W, n = 0/10) at day 14. However, there were no correspondences of abnormal AST to ALT values in these animals.
A transient reduction in platelet counts was observed at day 2 for the 100 μg pbi-shSTMN1-LP–treated group (n = 9/9), which returned to within normal range at later time points. Complete blood counts were otherwise within the normal reference range and did not significantly differ in any of the treatment groups.
Similarly, there were no consistent and remarkable histological changes to indicate treatment-related adverse events, based on microscopic examination of potential target organs (lungs, heart, kidneys, and skeletal muscle) as identified by previously conducted biodistribution studies (Templeton et al., 1997). Gross and histological changes identified in nontarget organs were sporadic, were infrequent, and could not be attributed to treatment or a particular dose. Thus, systemic injection of pbi-shSTMN1-LP fell within established safety tolerance for the observed time points. These cumulative gross toxicity and histopathologic findings indicate that the maximum tolerated dose (MTD) for administration of a single IV pbi-STMN1 LP injection was ≤100 μg (mouse equivalent of <26.5 μg; human equivalent dose (HED) of <0.09 mg/kg).
Discussion
RNAi technology has fueled the quest for personalized cancer therapy with its potential for high-throughput target identification and therapeutic application. There are currently at least 14 programs in various phases of clinical testing that are using RNAi methodology to achieve gene inhibition for various disease conditions (Vaishnaw et al., 2010). Among them, hepatic and extra-hepatic cancers (three programs) and chronic myelogenous leukemia (CML) (one program) are being targeted via systemic siRNA administration.
As a parallel RNAi-based technology, we have produced an expression plasmid–based bifunctional shRNA construct specific for STMN1, which is comprised of dual stem-loop structures to simultaneously generate two functionally distinct effector small RNAs (Wu et al., 2008). The pbi-shSTMN1 uses a miR30 backbone to navigate the intrinsic miRNA biogenesis process (Zeng et al., 2005) and is cloned into an expression plasmid under the control of an RNA pol II promoter (Lagos-Quintana et al., 2003; Lee et al., 2004). One shRNA contains matched stem sequences to promote Ago2-mediated passenger-strand cleavage, and the second shRNA has a partially mismatched stem sequence to promote cleavage-independent passenger-strand departure. Thus, functionality of the effectors is set by programmed passenger-strand–guided RNA induced silencing complex (RISC) loading rather than Ago subset distribution in the cancer cell. pbi-shSTMN1 achieved effective target knockdown with a significant dose advantage in tumor cell killing in vitro when compared with conventional STMN1-targeting shRNAs and siRNAs. Silencing of target gene expression was more effective, with a more rapid onset and greater durability of effect (Rao et al., 2010).
An effective gene-based therapeutic approach requires a platform that is effective and specific for systemic delivery of therapeutic doses of the agent to primary and metastatic tumor foci. Our liposomal delivery system incorporates a manually extruded formulation of DOTAP and cholesterol (Templeton et al., 1997) that forms bilamellar invaginated vesicles (BIVs) and encapsulates the expression plasmid. At the systemic level, enhanced homing to the targeted organ site by BIV is attributed to the enhanced permeation and retention effect in inflammatory sites and solid tumors in which the vasculature, particularly the larger endothelial junction, is leaky (Tong et al., 2009). BIV-encapsulated nucleic acids form flexible nanoplexes of 200–450 nm that penetrate the capillary fenestra of the tumor microenvironment as well as other tight intercellular junctions by virtue of their pliability. BIV complexes that are 200–450 nm in size have attained the highest levels of gene expression documented in all tissues and organs post–IV injections in mice (Ramesh et al., 2001; Templeton, 2002, 2009). Further, the liposomal BIV complexes are fusogenic, thereby bypassing endocytosis-mediated DNA cell entry that otherwise leads to nucleic acid degradation (Simberg and Barenholz, 2005) and TLR-mediated off-target effects. Patients with end-stage lung cancer have been given multiple IV infusions of DOTAP/cholesterol 3p FUS1 gene therapy without toxicity (Lu et al., 2007), and a unique DOTAP/cholesterol GNE gene lipoplex has been delivered via intramuscular and IV injection to a patient with hereditary inclusion body myopathy (HIBM2), a rare autosomal recessive neuromuscular disorder (Jay et al., 2008; Phadke et al., 2009; Nemunaitis et al., 2010).
Previously, Mistry and coworkers have demonstrated that ribozyme- and siRNA-based STMN1 posttranscriptional knockdown reduced human prostate cancer cells in vitro (Mistry et al., 2007; Mistry and Atweh, 2006) and osteosarcoma tumorigenicity in vivo (Wang et al., 2007). Our present observations of in vivo STMN1 knockdown and growth inhibition by pbi-STMN1 extend these findings to CCL-247 colorectal cancer xenografts, illustrating the applicability of this approach to preexisting human tumors.
With respect to the CCL-247 model, the benefit of repeat injections likely stem from introduction of pbi-shSTMN1 to previously untransfected tumor cells, given that an ∼5-fold increase in dose (from 7 to 34 μg) did not increase antitumor outcome. We observed marked STMN1 protein reduction in the treated tumor xenografts at 24 h after the last IT injection, confirming functionality of the pbi-shSTMN1-LP. Similar to our protein knockdown results, Huang et al. observed significant claudin-3 protein reduction at 3 days after the fourth IT injection (day 13 after first IT injection) of lipidiod/CLDN3 siRNA oligonucleotides into ovarian xenografts (Huang et al., 2009). A detailed, separate study with earlier time-point harvests and comprehensive analysis also involving in situ IHC analysis as well as global molecular analysis to adequately address the causal relationship of target gene knockdown with tumor reduction is planned.
The observed in vivo antitumor efficacy of pbi-shSTMN1 against tumorgraft models is instructive at several levels. Our positive treatment outcome in melanoma and osteosarcoma xenografts confirms the pivotal roles of STMN1 in the pathophysiology of these tumor cell types (Clauser et al., 1995; Watters et al., 1998; Wang et al., 2007). The significantly higher level of growth reduction with injection doses from 40 to 46 μg for these two models, not seen with the CCL-247 xenografts, may be related to a higher dependence of STMN1-related pathways in melanoma and osteosarcoma tumorgrafts (Clauser et al., 1995; Watters et al., 1998; Wang et al., 2007). The markedly reduced viability of residual tumor mass following STMN1 knockdown confirmed our earlier in vitro findings and suggests that efficacy based on tumor size measurements may actually underestimate the growth inhibitory outcome of this treatment approach analogous to reported observations in the clinical ONYX-015 trials (Reid et al., 2005).
A predominance of necrotic tissues within residual tumors is an explanation for the extended tumor-static findings of up to day 46 in the low-passage melanoma tumorgrafts. By comparison, duration of tumor reduction by siRNA oligonucleotide lipoplexes or naked siRNA were reported to be 4 days after second IT injection and 3 days after the last (fourth) injection in murine breast cancer syngeneic 4T1 (Tsutsumi et al., 2009) and ovarian xenograft (Huang et al., 2009) models, respectively. Thus, tumor growth reduction that was extended up to day 25 constituted a markedly extended treatment outcome by our bifunctional plasmid therapeutic. Correspondingly, we have demonstrated mature shRNA in pbi-shSTMN1-LP–treated low-passage osteosarcoma tumorgrafts for up to day 16 after the last IT injection based on a previously validated RT-qPCR method for miRNA (Chen et al., 2005) when compared with the detectable duration of 7 days for siRNA oligonucleotides by a similar assay (Abrams et al., 2010). These findings support the clinical relevance of the pbi-shSTMN1 approach, given evidence that heterotransplanted primary human tumors better resemble their clinical counterparts with respect to their biomolecular features and responsiveness to treatment, including the metastatic subset (Fu et al., 1992; Perez-Soler et al., 2000; Belletti et al., 2008; Lopez-Barcons, 2009; Revheim et al., 2009).
We performed sequential studies to demonstrate the feasibility of growth inhibition with a single injection and then to establish optimal treatment outcome with repeat injections. The initial repeat injection schemes of three or six daily injections were based on previous data by Ito et al. (2004) and Ramesh et al. (2001). Both utilized a similar DOTAP:cholesterol formulation to successfully deliver therapeutic transgenes and achieved the desired tumor growth inhibition effect. However, recent studies with liposomal (Ryschich et al., 2007) or polyethylenimine (Li et al., 2007; Kang et al., 2009) delivery or naked siRNA (Pille et al., 2005) have generated similar antitumor activity by administering IT injections two or three times weekly (three to seven total injections). On the basis of results obtained from the melanoma and osteosarcoma tumorgraft studies, our schedule of six injections given semiweekly is considered to be the regimen of choice.
Our findings with pbi-shSTMN1-LP in biorelevant rats indicates that this nanotherapeutic is safe and well tolerated via both single or multiple IT and single IV administration. The IT MTD of >92 μg in immune-compromised mice (HED of 0.3 mg/kg) was ∼10-fold higher than the extrapolated IC50 (9 μg, HED of 0.03 mg/kg) in the single IT treatment study with CCL-247 tumor xenografts. pbi-shSTMN1-LP demonstrated a dose-dependent efficacy, exerting maximal effect at a dose of 7–8 μg (CCL-247 tumor xenograft model) and 46 μg (low-passage melanoma tumorgraft model), respectively. At the systemic level, toxicology studies showed that the expected therapeutic dose can be safely administered, with an MTD of ≤100 μg (mouse equivalent of <26.5 μg; HED of <0.09 mg/kg). Repeat IV injections with a similar LP construct to deliver PDX-1 oncogene-specific shRNAs have similarly led to highly effective tumor growth inhibition and survival advantage in a SCID-hu pancreatic cancer model (Liu et al., 2008). Hence, the feasibility of repeat administrations with the BIV lipoplex formulation can be harnessed to achieve increased transfection efficiency and/or overcome tumor barriers (Sinek et al., 2009).
Conclusion
In conclusion, we have provided proof-of-principle efficacy studies that demonstrate the feasibility of using bifunctional RNAi platform for reducing STMN1 gene expression in vivo. Continuing studies aim to improve this platform in justification for targeted systemic delivery to malignant-involved areas. We have previously published successful targeted delivery of DNA products with the current delivery vehicle (Shi et al., 2010) and have demonstrated successful systemic knockdown effect and survival advantage with bifunctional RNAi of other targets (i.e., PDX-1) with the same delivery vehicle in xenograft models (unpublished data). Our IT studies have identified the appropriate therapeutic dose for pbi-shSTMN1. Preclinical toxicology data using the bifunctional RNAi platform adds pertinent safety data to the rationale for performing IT phase I studies in cancer patients to confirm safety, document tumor effector uptake and STMN1 downregulation, and determine the phase II recommended dose.
Footnotes
Acknowledgments
The authors thank Kimberly Winterrowd and Kimberly Cavanaugh at UNT Health Science Center for their assistance with animal studies. The authors gratefully acknowledge the generous support of the W.W. Caruth Jr. Foundation Fund of the Communities Foundation of Texas, the Jasper L. and Jack Denton Wilson Foundation, and the Merkle Family Foundation. The authors also thank Susan Mill and Brenda Marr for their competent assistance in the preparation of this article.
Disclosure Statement
The following authors are shareholders in Gradalis, Inc.: John Nemunaitis, Neil Senzer, Phillip Maples, Donald Rao, and Nancy Templeton. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in this article.