
Abstract
Toll-like receptor 4 (TLR4) responds to lipid A, the active moiety of lipopolysaccharide from Gram-negative bacteria, in cooperation with myeloid differentiation protein-2 and plays a vital role in innate immunity. Polymorphisms in TLR4 are associated with changes in susceptibility to various infectious diseases. We previously found seven amino acid polymorphisms in Sus scrofa TLR4. In this study, we showed by luciferase reporter assay that an alteration from cysteine to tryptophan at position 506 (C506W) caused loss of ability to induce nuclear factor-κB activation after lipid A stimulation. This polymorphism was found only in Japanese wild boar (JWB) populations of S. scrofa. Genotyping of TLR4 in different JWB populations revealed that C506W polymorphism was under pressure from purifying selection in a local population (Tajima's D=−0.98; p<0.05). However, in another population, this polymorphism existed at a frequency such that homozygous animals with the W506 alleles seldom appeared. These findings suggest that the C506W polymorphism is under different types of pressure by natural selection between populations, which may reflect differences in residential pathogens or demographic factors.
Introduction
Many studies have shown that polymorphisms in human TLR4 are associated with increased resistance and susceptibility to various diseases (Lazarus et al., 2002; Schröder and Schumann, 2005). The D299G polymorphism in human TLR4 is associated with increased mortality in septic shock (Lorenz et al., 2002); conversely, the presence of this allele is associated with decreased mortality from severe malaria (Mockenhaupt et al., 2006). Ferwerda et al. (2007) demonstrated that the D299G allele and the D299G/T399I haplotype are concentrated differently in African and European populations, respectively. In addition, evidence that strong purifying selection operates on TLR4 was obtained in an Indian population, although no significant evidence of selection pressure was found in American populations, who live in regions with lower microbial loads than those found in India (Mukherjee et al., 2009). These findings imply that habitat—especially the presence of specific pathogens in a local area—determines the natural selection pressure exerted on the TLR4 gene.
In the swine industry, opportunistic infections such as pneumonia and diarrhea are major problems and lead to extensive economic losses (Straw et al., 2006). Polymorphisms in TLR4 are expected to be associated with changes in susceptibility to these infectious diseases in swine. We previously searched for single-nucleotide polymorphisms (SNPs) in TLR4 among Sus scrofa consisting of 96 individuals from 11 breeds and detected seven SNPs that resulted in amino acid substitutions (Shinkai et al., 2006). Of these polymorphisms, L204H, R321H, and Q343K were frequently detected among 259 animals belonging to commercial and traditional European swine populations (Palermo et al., 2009), whereas N189H, V276I, S386N, and C506W were detected in only a few animals in our previous study (Shinkai et al., 2006). Here, by using a luciferase reporter assay, we found that one of these substitutions, an alteration from cysteine to tryptophan at position 506 (C506W), which was detected only in Japanese wild boar (JWB) populations, absolutely impaired LPS signaling. In addition, genotyping of the C506W polymorphism revealed the restricted distribution of this SNP among different JWB populations. These results suggest that the malfunction allele of TLR4 is disappearing from JWB populations under pressure from purifying selection, although the allele remains at a certain frequency in a specific population potentially because of differences in residential pathogens or demographic factors.
Materials and Methods
Cell and reagents
Human embryonic kidney (HEK) 293 cells (American Type Culture Collection [ATCC] CRL-1573) were maintained in a 5% CO2 incubator at 37°C in Dulbecco's modified Eagle medium (Gibco/Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Gibco/Invitrogen). Lipid A from Escherichia coli serotype R515 was purchased from Alexis Biochemicals.
Expression vectors
The coding regions of the S. scrofa TLR4 and LY96 (encoding myeloid differentiation protein-2 [MD-2]) genes were amplified by polymerase chain reaction (PCR) with plasmid DNA from an alveolar macrophage cDNA library (Uenishi et al., 2007) and with total RNA derived from the thymus of a 1-month-old male Landrace breed pig, respectively, with primers composed of the most common nucleotide sequences in European pigs (TLR4, GenBank ID: AB188301; LY96, GenBank ID: AB267811). PCR amplification was conducted by using PfuTurbo DNA polymerase (Stratagene) with 40 cycles, each consisting of 95°C for 30 s, 60°C for 30 s, and 72°C for 2 min, after incubation at 95°C for 2 min. TLR4 reference and MD-2 expression vectors were constructed by inserting the coding regions into pEF6/V5-His TOPO (Invitrogen), as previously described (Bochud et al., 2003). Expression vectors encoding mutant TLR4 proteins were produced by introducing each of the seven SNPs into the reference vector just mentioned by using a QuikChange Site-Directed Mutagenesis Kit (Stratagene) in accordance with the manufacturer's instructions. A nuclear factor-κB (NF-κB)–dependent firefly luciferase construct was generated as previously described (Shinkai et al., 2011). Thymidine kinase Renilla luciferase vector (pGL4.74; Promega) was used as a control.
Luciferase reporter assay
HEK293 cells were transiently cotransfected, using FuGENE 6 Transfection Reagent (Roche Diagnostics), at a density of 6×104 cells per well in a 96-well plate format, with 50 ng of one or two of the TLR4 expression vectors, 50 ng MD-2 expression vector, 100 ng NF-κB–dependent firefly luciferase construct, and 20 ng thymidine kinase Renilla luciferase vector. After 24 h, the cells were stimulated with 50 ng/mL of lipid A for 5 h. Luciferase activity was measured with a Dual-Glo Luciferase Assay System (Promega). After the firefly luciferase units had been divided by the Renilla units, the values of stimulated cells were divided by those of unstimulated cells to show the degree of induction of NF-κB. All experiments were performed three times in triplicate wells to confirm reproducibility.
Western blot
HEK293 cells were transiently transfected with 55 μg each of EF6 control, TLR4 reference, and TLR4-C506W expression vectors by using FuGENE 6 at 1×107 cells per 75-cm2 flask. After 24 h, cells were washed twice with cold phosphate-buffered saline and treated with a Cell Surface Protein Isolation Kit (Pierce), in accordance with the manufacturer's instructions, to isolate Biotin-labeled cell surface proteins. Briefly, for biotinylation of cell surface proteins, cells were incubated with 0.25 mg/mL Sulfo-NHS-SS-Biotin (Pierce) for 30 min at 4°C. After the reaction had been quenched, cells were washed twice with Tris-buffered saline and lysed with lysis buffer in the presence of protease inhibitors (Halt Protease Inhibitor Cocktail Kit; Pierce). Biotinylated proteins were immunoprecipitated with NeutrAvidin-agarose (Pierce) while being rotated for 60 min at room temperature; they were then eluted in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sample buffer (Lane Marker Sample Buffers; Pierce). The concentrations of eluted proteins were determined with a Micro BCA Protein Assay Kit (Pierce). One microgram each of the proteins was separated by using SDS-PAGE, transferred to a polyvinylidene difluoride membrane by using an iBlot Dry Blotting System (Invitrogen), and immunoblotted with Anti-V5-HRP Antibody (Invitrogen).
Analyses of molecular population genetics
Tissue or blood samples of JWB populations in Gunma and Shizuoka Prefectures and also in 10 additional regions of Japan were purchased from hunters with legal permission for hunting wild boars. The samples were used in this study following Japanese laws and the rules of our institutes. Genomic DNA from these samples was extracted using a standard phenol–chloroform protocol (Sambrook and Russell, 2001). DNA sequencing and subsequent detection of SNPs in the entire coding region of TLR4 were conducted in samples from 96 boars in Gunma Prefecture and 96 in Shizuoka Prefecture as previously described (Shinkai et al., 2006). Using Arlequin 3.5 software (Excoffier et al., 2005), the expectation-maximization algorithm (Excoffier and Slatkin, 1995) was used to estimate haplotypes from genotypic data for which the gametic phase was unknown. Haplotype phases were determined for 177 and 159 alleles in the populations from Gunma and Shizuoka Prefectures, respectively, excluding alleles with poor-quality genomic DNA or sequence traces. Calculation of mean numbers of differences in nucleotides between all pairs of haplotypes and Tajima's test for selective neutrality were performed by using Arlequin 3.5 software. The test statistic D was computed by comparing the observed frequencies of variants at polymorphic sites to the frequencies expected under neutral theory (Tajima, 1989). The significance of the D statistics was calculated by generating 1000 random samples (Excoffier and Lischer, 2010). Haplotype diversity (h) was calculated by using the following formula: h=n(1–Σxi 2)/(n–1), where n is the number of samples, and xi is the frequency of the ith haplotype (Nei, 1987).
Sequence alignment and homology modeling
Comparison of amino acid sequences of TLR4 of vertebrates was conducted using ClustalX2 (Larkin et al., 2007). The three-dimensional (3D) structure of the ectodomain of porcine TLR4 was constructed by using MODELLER Release 9v8 (Šali and Blundell, 1993). The crystal structure of the dimer of human TLR4–MD-2–LPS complex (PDB ID: 3FXI) was used as a template (Park et al., 2009). The structures were then visualized by using a combination of the PyMOL Molecular Graphics System (Version 1.3; Schrödinger) and POV-Ray (Version 3.6; Persistence of Vision Pty. Ltd.).
Results
Function of TLR4 impaired by C506W amino acid polymorphism
To study the effect on lipid A recognition produced by seven amino acid substitutions within the ectodomain of S. scrofa TLR4 (Shinkai et al., 2006) (Fig. 1a), we performed a NF-κB luciferase reporter assay using HEK293 cells cotransfected with LY96 (MD-2), which encodes an accessory molecule associating with the ectodomain of TLR4 (Shimazu et al., 1999), and a TLR4 mutant vector carrying one of the nonsynonymous SNPs. On stimulation with lipid A, transfectants with the N189H, L204H, R321H, or Q343K mutant showed levels of NF-κB induction similar to the level of the TLR4 reference (Fig. 1b). Those with V276I or S386N could respond to lipid A but showed a significant and slight decrease in induction (p<0.01 by Welch's t-test). In contrast, the transfectant with the C506W mutant exhibited absolutely impaired response to lipid A stimulation, as did the EF6 control (p<0.005 by Welch's t-test). Although the expression of TLR4 protein from the C506W mutant vector was relatively low compared with that of the reference vector, the low amount of protein could not explain the full abrogation of the function by the mutation (Fig. 1c). These results indicated that the C506W polymorphism completely impaired LPS signaling of TLR4 probably because of adverse effects on the conformation of the protein. The C506W polymorphism did not appear to have a dominant-negative effect based on the result of the reporter assay in which the TLR4 reference and C506W mutant vectors were concurrently transfected (Fig. 2a).
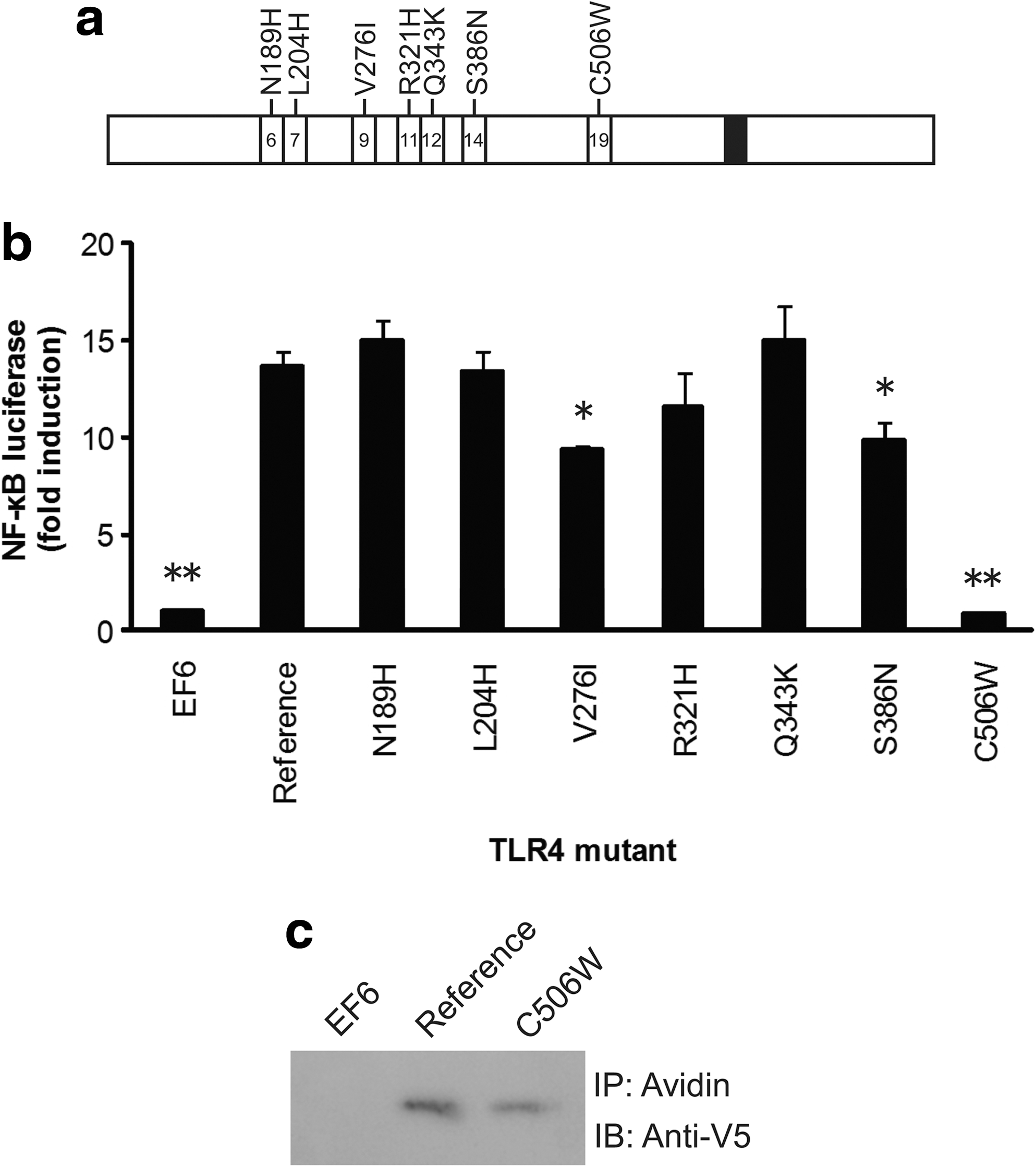
Amino acid polymorphisms in Sus scrofa TLR4 and their influence on the recognition of lipopolysaccharide.
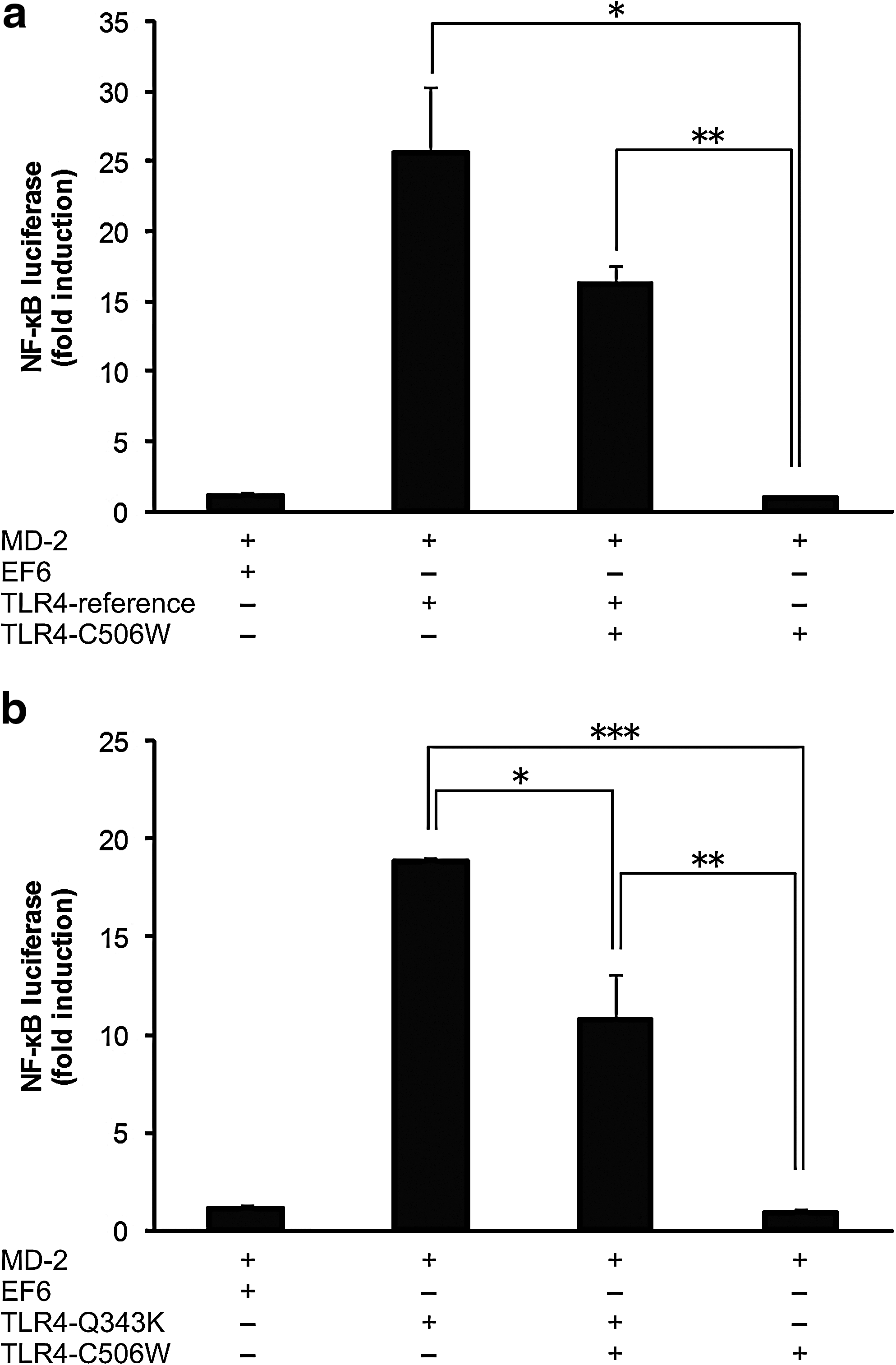
Influence of the combination of different TLR4 mutant vectors on the recognition of lipid A. HEK293 cells were transiently cotransfected with 50 ng MD-2, and 50 ng EF6 or TLR4 expression vectors as indicated, together with NF-κB reporter and Renilla control vectors. For double cotransfections with TLR4-reference and TLR4-C506W
Sequence diversity of the TLR4 gene in JWB populations
In our previous study (Shinkai et al., 2006), an amino acid alteration of C506W in the 19th LRR motif (Fig. 3), resulting from an SNP of thymine to guanine at position 1518 (T1518G) in the coding sequence of TLR4, was heterozygously found in only one JWB individual among five. To examine the distribution of this SNP and the possibility of unknown polymorphism in S. scrofa TLR4, we sequenced the coding region of the gene in two sets of genomic DNA from JWB populations. We found six SNPs, consisting of five SNPs previously detected, at nucleotide positions 318, 960, 962, 1027, and 1518 (Shinkai et al., 2006) and a new SNP of cytosine to thymine at position 282 (Table 1). Estimation of haplotypes from the genotypic data revealed six and five haplotypes in the populations from Gunma and Shizuoka Prefectures, respectively (Table 1). In the population from Gunma, the haplotype including the guanine allele at position 1518 was present at a frequency of about 10%, despite the absolute disadvantage in LPS signaling (Fig. 1b). In contrast, the frequency of this haplotype was very rare in the population from Shizuoka. The haplotype including the cytosine allele at position 1027 was exclusively observed in the population from Gunma. Therefore, the amino acid polymorphism of Q343K (C1027A SNP) could compensate for the deleterious effect of C506W (T1518G SNP) in animals with the respective minor alleles at both SNP sites. However, the result of the reporter assay in which these two mutant vectors had been concurrently cotransfected did not show any additional effect of Q343K in combination with C506W (Fig. 2b). The average pairwise nucleotide diversity per site (π) (Nei, 1987) was 5.8×10−4 in the Gunma population and 2.1×10−4 in the Shizuoka population. In addition, the haplotype diversity (h) was 0.69 in the Gunma population and 0.33 in the Shizuoka population. These results indicate that the diversity of TLR4 differs widely in the local populations of JWB.
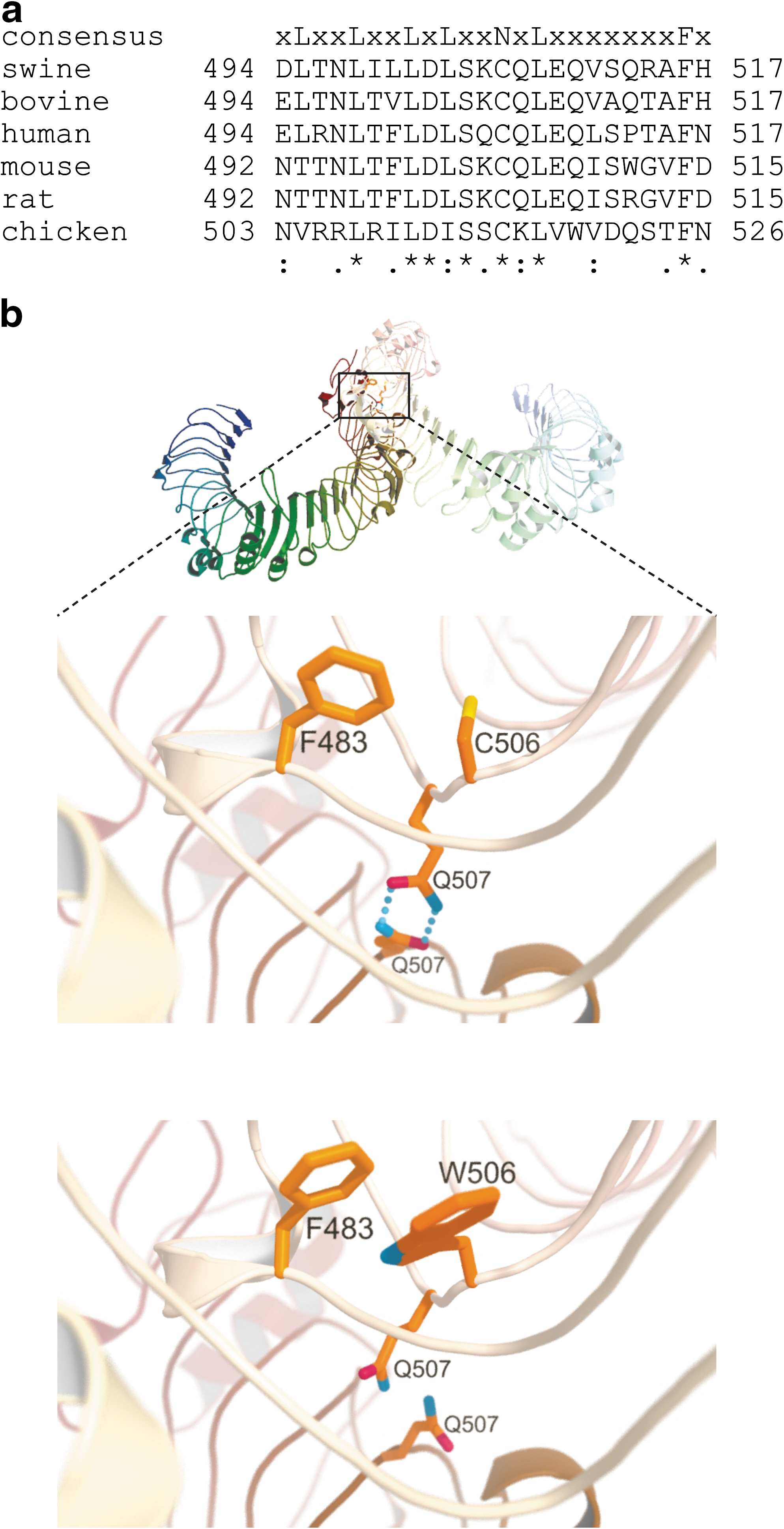
Location of the C506W polymorphism within the TLR4 protein.
Haplotypes consist of six SNPs; nucleotides that are identical to those of the major haplotype (CCAAAT) are indicated by dashes. SNPs at nucleotide position numbers 282, 318, and 960 were synonymous substitutions, and those at positions 962, 1027, and 1518 were nonsynonymous substitutions, which caused amino acid alterations of R321H, Q343K, and C506W, respectively. SNP, single-nucleotide polymorphism.
Tests of neutrality for TLR4 in JWB populations
To detect whether polymorphisms within TLR4 in the JWB populations were under natural selection, we calculated Tajima's D (Tajima, 1989) by using 177 alleles in the Gunma population and 159 in the Shizuoka population. D values estimated for the entire coding region of TLR4 were 0.818 (p=0.832) in the Gunma population and −0.818 (p=0.234) in the Shizuoka population. These values imply that the apparent pressures of balancing or purifying selection were not exerted on the TLR4 gene in either population. However, analysis of a sliding-window plot of D suggested that the regions containing the SNPs of C318A (D=−0.977; p=0.061) and T1518G (D=−0.977; p=0.046), respectively, were under pressure of purifying selection in the Shizuoka population (Fig. 4). In contrast, in the Gunma population, the region containing the SNP of C318A was potentially under pressure of purifying selection (D=−0.965; p=0.047), whereas the region containing the SNPs of A960G and A962G showed positive D statistics (D=2.056; p=0.977), thus suggesting that gene flow into the Gunma population had recently occurred. The region containing the SNP of T1518A was not under pressure of natural selection in the Gunma population (D=0.058; p=0.733), despite the disadvantage in LPS signaling (Fig. 1b).
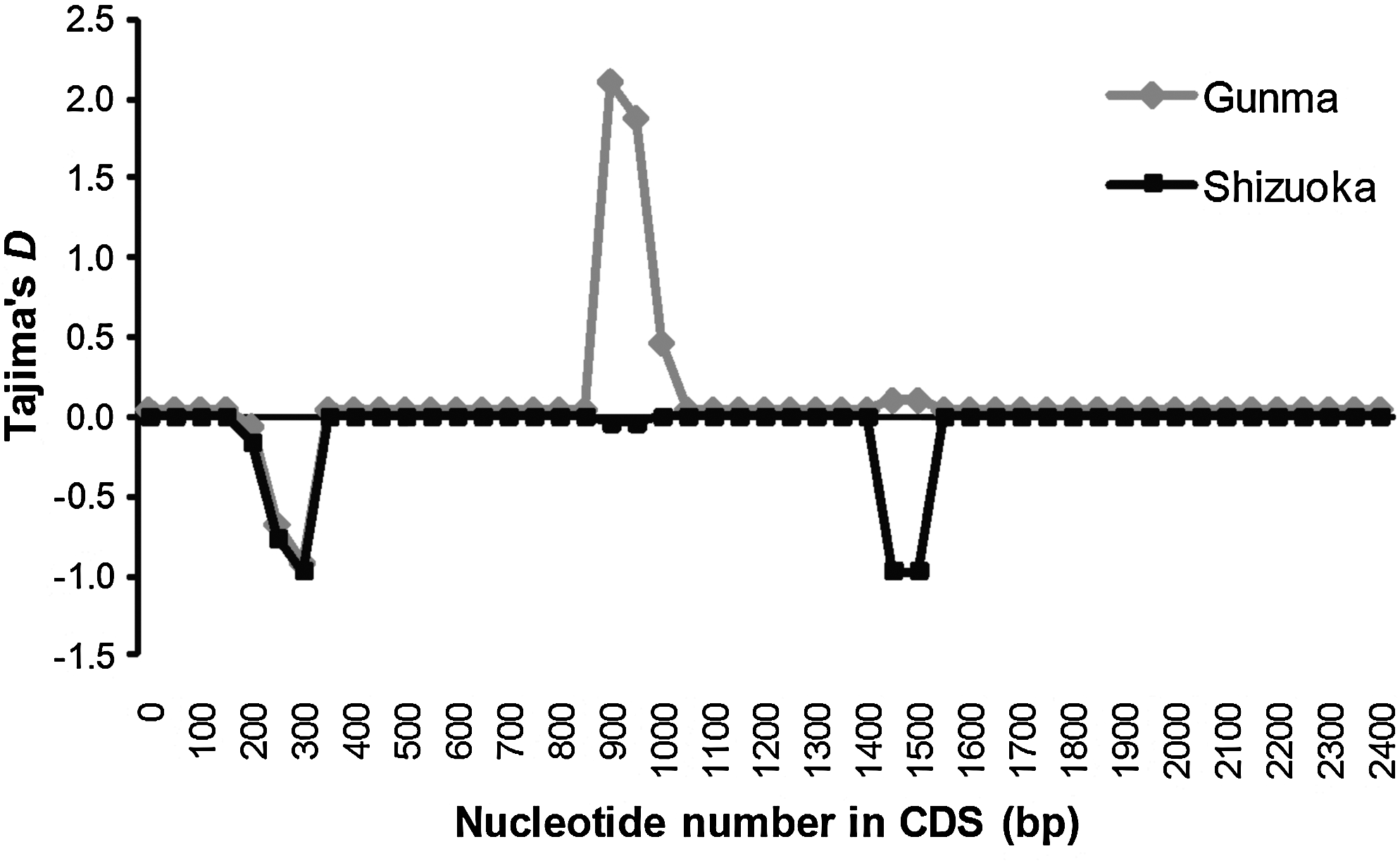
Sliding-window plot of Tajima's D values across the coding nucleotide sequence (CDS) of TLR4 in Japanese wild boar populations. Values were calculated within every 100-bp window, sliding by 50 bp, and are indicated as gray and black squares for local populations from Gunma and Shizuoka prefectures, respectively.
Discussion
We found by luciferase reporter assay that the amino acid polymorphism of C506W within S. scrofa TLR4 absolutely impairs LPS signaling. Our molecular population genetic analysis indicates that the responsible SNP is influenced by different types of pressure by natural selection between distinct JWB populations.
The mechanism of LPS recognition by TLR4 has been extensively studied. TLR4 requires association with MD-2 for the recognition of LPS (Shimazu et al., 1999). LPS from Gram-negative bacteria is transferred to the TLR4–MD-2 complex by two accessory proteins, LPS-binding protein and CD14 (Miyake, 2006). This binding of LPS induces aggregation of the TLR4–MD-2 complex and initiates intracellular signaling (Kobayashi et al., 2006). Examination of the crystal structures of the TLR4–MD-2 complex has elucidated the amino acids and regions of TLR4 involved in the interaction with MD-2 and LPS. The N-terminal region of TLR4, containing amino acids at positions 41 to 288, is responsible for the interaction with MD-2 via charge-enhanced hydrogen bonds (Kim et al., 2007). In addition, examination of the dimer structure of the TLR4–MD-2–LPS complex has revealed that the region containing amino acids at positions 416 to 463 within TLR4 in one TLR4–MD-2 complex participates in the dimerization interface with MD-2 in the other complex (Park et al., 2009). LPS is anchored by seven amino acids in the region covering positions 264 to 463 of TLR4 (Park et al., 2009). These physical relationships suggest that the cysteine residue at the 506th amino acid within TLR4, which is highly conserved in the 19th LRR unit among vertebrates (Fig. 3a), is unlikely to be involved in direct interaction with MD-2 and LPS. In contrast, amino acid residues N365, S386, V411, N433, and Q507 compose the homodimerization interface of TLR4 (Park et al., 2009). Together with the speculated burial of C506 within the 3D structure (Fig. 3b), alteration of cysteine to tryptophan can be expected to change the conformation of the TLR4 protein, especially in the case of the side chain of Q507. This hypothesis is supported by the fact that the tryptophan residue has a large indole ring at the side chain, and a phenylalanine residue is present in the neighborhood of the 506th residue (Fig. 3b). This conformational change in the TLR4 protein is expected to influence the dimerization of the TLR4–MD-2–LPS complex.
In this study, we examined the efficiency of NF-κB induction for seven TLR4 mutant proteins after stimulation with lipid A using a luciferase reporter assay. The polymorphisms L204H, R321H, and Q343K, which were the common SNPs detected in our sample (Shinkai et al. 2006), and also in the European samples studies by Palermo et al. (2009), showed levels of NF-κB induction comparable to the level of the TLR4 reference (Fig. 1b). In contrast, the rare polymorphisms V276I, S386N, and C506W (though not N189H), which were observed only in our samples, showed significantly lower levels of NF-κB induction compared with the level of the reference. These results are consistent with the neutral theory of molecular evolution that neutral mutations are widespread in populations as a result of genetic drift (Kimura, 1989). On the other hand, mutations that impair gene function will likely disappear from populations.
However, the haplotype including the W506 allele, which completely impaired the function of LPS signaling (Fig. 1b), remained at a frequency of about 10% in the population of JWB from Gunma, although it was disappearing in the population from Shizuoka (Table 1). In a previous study, we genotyped T1518G—the SNP responsible for the C506W—in various populations of swine (Shinkai et al., 2006). In addition, we have genotyped this SNP in more than 100 domestic pigs (S. scrofa domesticus) and 288 JWB individuals from several regions other than Gunma and Shizuoka Prefectures of Japan (Supplementary Table S1; Supplementary Data are available online at
On the other hand, the C1027A SNP could complement the deleterious effect of T1518G, because both SNPs were observed almost exclusively in the Gunma population (Table 1). However, the C1027A SNP did not show any additive effect in combination with the T1518G SNP in the in vitro reporter assay (Fig. 2b). Therefore, the distribution of the T1518G SNP in the population from Gunma seems to be unaffected by the mutant protein from the C1027A SNP; however, the possibility that other factors (e.g., the expression efficiency of the promoter region linked to the C1027A SNP) may have some effects cannot be excluded.
The TLR4 allele with the C506W polymorphism was almost exclusively observed in the Gunma population. The origin of this malfunctioning TLR4 allele is interesting. The haplotype carrying nucleotide polymorphisms G960, G962, and C1027 is found frequently in European pig breeds (Palermo et al., 2009), but has not been found in JWB animals, except for the Gunma population (Table 1). This suggests that the gene flow from domestic pig(s) into the Gunma JWB population is a recent occurrence, a concept that is supported by the high Tajima's D statistics around the genomic region carrying the three SNPs. In contrast, the D value of the region around the T1518G nucleotide polymorphism (C506W) was approximately zero, and T1518 did not have linkage with A960G, A962G, or C1027A, thus suggesting that the origin of T1518G is different from that of the three SNPs. The inability to detect T1518G in pigs and its highly biased distribution among JWB populations implies that the T1518G SNP originally emerged in a Gunma JWB animal or that it originated in an ancestor of S. scrofa and has remained in the Gunma population.
Although Gunma and Shizuoka Prefectures are geographically close (about 60 km apart), they are separated by Japan's highest mountains, including Mt. Fuji, and by cities with large populations. Hence, interaction between JWB in the two prefectures is likely to be difficult. In addition, the climate differs substantially between the two prefectures, and this may also cause the distributions of pathogens to differ. This difference in residential pathogens may be the cause of the different selection pressures exerted on TLR4 between populations from Gunma and Shizuoka Prefectures. Further study of residential pathogens and polymorphisms of genes other than TLR4 in various regions of Japan may elucidate the origin of the unique distribution of the T1518G SNP in the population from Gunma Prefecture. We previously reported that the amino acid polymorphisms TLR2V703M, TLR5R148L, and TLR5P402L in swine attenuate the response to Salmonella enterica serovar Choleraesuis and that each polymorphism was observed only in Berkshire, Jinhua, and Landrace breeds, respectively (Shinkai et al., 2011). These biased distributions may be influenced by regionally specific pathogens or demographic factors as well as by domestication by humans.
Footnotes
Acknowledgments
This work was supported in part by the Animal Genome Research Project of the Ministry of Agriculture, Forestry, and Fisheries of Japan, and by a Grant-in-Aid from the Japan Racing Association.
Disclosure Statement
The authors declare that there is no conflict of interest in this study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.