
Abstract
Caspase-2 is an enigmatic caspase that is now increasingly being associated with certain types of cell death in cells exposed to cytotoxic agents. It is now known that in some cases of cell stress, such as DNA damage, activation of this caspase is triggered, sometimes in the absence of activation of both the intrinsic and extrinsic pathways of apoptosis. Part of the reason for this enigma has been lack of a suitable stimulus for this caspase, and with the discovery of DNAzyme 13 (Dz13), a potent oligonucleotide-based caspase-2 activator, much more can now be elucidated. For instance, one thing that could be unraveled is whether caspase-8 and Fas (CD95)-associated protein with death domain are indeed involved in caspase-2 activation as part of the death-inducing signaling complex. It is also becoming apparent that this enigmatic caspase may be important in the mechanisms behind which chemotherapeutic agents inhibit tumor cell growth. A better understanding of the true biological effects of this enzyme may indeed lead to more effective ways of managing tumors clinically. This review article briefly examines the different compounds capable of inducing activation of caspase-2 and proposes Dz13 as one that will be valuable for evaluation of the biological functions of caspase-2.
Introduction
PIDD, p53-induced protein with a death domain; Dz13, DNAzyme 13.
Recently, it has been reported that loss of caspase-2 results in an increased ability of cells to acquire a transformed phenotype and gain malignancy, suggesting that caspase-2 is a tumor suppressor protein (Ho et al., 2009). Also, it has been shown that caspase-2 leads to apoptosis by suppression of the survivin gene, thereby acting as an endogenous inhibitor of nuclear factor-kappa B–dependent cell survival and this mechanism may contribute to tumor suppression in humans (Guha et al., 2010). Caspase-2 activation has been noted to permeabilize mitochondria, and p53-induced protein with a death domain (PIDD) and RIP-associated Ich-1/CED homologous protein with death domain (RAIDD) are important for caspase-2 activation (Tinel and Tschopp, 2004; Baptiste-Okoh et al., 2008a, 2008b), though not in all cases (Vakifahmetoglu et al., 2008; Manzl et al., 2009). PIDD is dispensable for both p53-dependent and -independent apoptosis (Kim et al., 2009). It is notable that caspase-2 activation can be repressed by p21 (Baptiste-Okoh et al., 2008a), and in turn, p21 expression is regulated by caspase-2 at the translational level (Sohn et al., 2011). Figure 1 demonstrates the caspase-2–mediated apoptotic pathway.
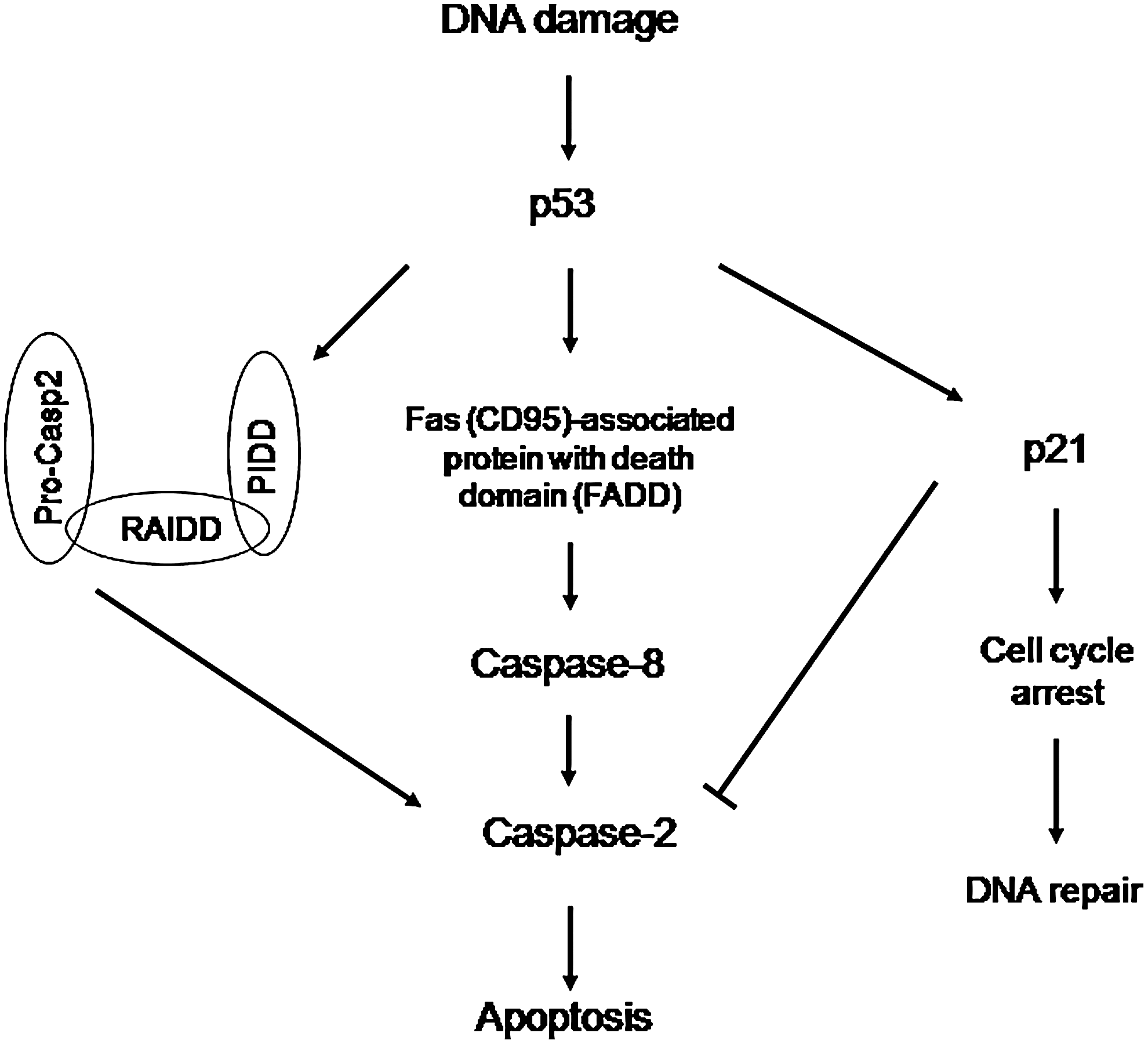
Caspase-2–mediated apoptotic pathway induced by DNA damage. In this model, central to the cell's fate is whether caspase-2 is upregulated or downregulated. DNA damage induces p53 activation, and in turn, p53 activates either PIDDosome or FADD, which results in caspase-2 activation. Or p53 transactivates p21, which downregulates caspase-2 and leads to cell cycle arrest and, in turn, results in DNA repair instead of apoptosis. Adapted from the works of Olsson et al. (2009) and Baptiste-Okoh et al. (2008a). PIDD, p53-induced protein with a death domain; RAIDD, RIP-associated Ich-1/CED homologous protein with death domain; FADD, Fas(CD95)-associated protein with death domain.
As aforementioned, of all caspases, and despite being one of the most conserved caspases across species, caspase-2 is one about which relatively little is known (Kitevska et al., 2009), and this is partly due to lack of a strong inducer for activation. Against a backdrop of such a deficiency, recent studies demonstrated that some agents, ranging from a widely used anticancer drug to a radiosensitizer, induce caspase-2 activation, and these findings are summarized in Table 2. In particular, a potent inducer of this caspase, an oligonucleotide called DNAzyme 13 (Dz13; a 34mer deoxyribozyme downregulating c-jun expression, Khachigian et al., 2002; Dass 2004), has been recently shown to activate caspase-2 potently in a variety of tumor cell lines (Dass and Choong, 2010). In this review, we discuss recent findings on caspase-2, focusing on chemotherapy-induced activation of this enzyme.
Induction of Caspase-2 Activation in Tumor Cells by Dz13
Previously, the deoxyribozyme Dz13 has been shown to reduce the growth of melanoma indirectly via antiangiogenesis (Zhang et al., 2004) and squamous cell carcinoma directly (Zhang et al., 2006). Dz13 downregulates c-jun (its specific target) levels in rapidly proliferating cells, for instance, when cells are stimulated postquiescence or when c-Jun is elevated (Bhindi et al., 2007). More recently, Dz13 has been shown to inhibit the growth of both osteosarcoma (OS) (Dass et al., 2008b, 2008c, 2008e) and liposarcoma (LS) (Dass et al., 2008d) in orthotopic models in our lab. Tumor growth is inhibited in orthotopic models of prostate cancer, breast cancer, and OS in bone of mice by Dz13 when tumor cells are mixed with Dz13 preimplantation (Tan et al., 2010c). Normal cells in bone such as osteoblasts and chondrocytes are left relatively unharmed, indicating a tumor cell–selective mode of action for Dz13 (Tan et al., 2010b). This finding was quite intriguing and suggested that a fundamental difference between tumor cells and normal cells was being exploited for such a differential cell kill response.
When Dz13-treated cells were observed with electron microscopy, death of cells occurred, exhibiting the classical signs of apoptosis (Tinari et al., 2008)—nuclear fragmentation, chromatin condensation, and membrane blebbing. In addition, mitochondrial structure was perturbed, with septae destroyed. Apoptosis was confirmed with poly (ADP-ribose) polymerase 1 (PARP-1) cleavage, terminal deoxynucleotidyl transferase dUTP nick and labelling (TUNEL) assay, annexin-5 staining, and nuclear chromatin condensation as evidenced by 4′,6-diamidino-2-phenylindole staining (Dass et al., 2010). Structures resembling transfection complexes were observed within cells, specifically within endosome-like organelles. Death occurred via the mitochondrial pathway as shown by cytochrome c release into the cytosol.
Dz13-mediated tumor cell death via apoptosis was determined to be due to caspase-2 activity, as chemical inhibitors to other caspases failed to abrogate Dz13-mediated cell kill (Dass et al., 2010a). The pan-caspase inhibitor (benzyloxycarbonyl-Val-Ala-Asp (OMe)-fluoromethyl ketone [z-VAD-FMK]) and caspase-2 inhibitor (benzyloxycarbonyl-Val-Asp (OMe)-Val-Ala-Asp (OMe) fluoromethyl ketone [z-VDVAD-FMK]) inhibited Dz13-mediated cell death, once again confirming that the mode of cell death was apoptosis. These results for caspase-2 were then confirmed when siRNA to caspase-2 also abrogated Dz13-mediated apoptosis. Finally, western blotting of cell lysates revealed a clear activation of caspase-2 from its 51 kDa band to its 33 kDa band.
To confirm that only caspase-2 was activated by Dz13 in tumor cells, western blotting was used to screen other effector caspases in the cell lines (Dass et al., 2010a), but none was activated. In addition, PIDD and RAIDD are closely associated with caspase-2 activation (Tinel and Tschopp, 2004). However, previous studies have shown that both PIDD and RAIDD are dispensable for caspase-2 activation (Vakifahmetoglu et al., 2008; Manzl et al., 2009), although, so far, only one report has demonstrated the significance of DNA-protein kinase catalytic subunit (PKcs) in caspase-2 activation. Hence, overall, research is very preliminary in this area of caspase-2-piddosome interaction. Although PIDD and RAIDD were activated or upregulated, respectively, when cells were treated with Dz13, all were deemed to be nonessential for Dz13 activity (Dass et al., 2010a, 2010b) when downregulation with their respective siRNAs failed to abrogate Dz13-mediated cell death.
As Dz13 activation of caspase-2 caused significant tumor cell death in vitro (Dass et al., 2010a), Dz13 was evaluated in orthotopic models of various tumors. Tumors (in this case, of the bone) were allowed to establish before treatment was initiated. These pre-established tumors resemble those observed when patients present to the clinic with initial complaints such as joint tenderness and/or swelling and are scheduled for an initial X-ray and/or magnetic resonance imaging (MRI). Both primary and secondary tumors are inhibited with chitosan-based nanoparticles encapsulating Dz13 (Dz13-NP; Dass et al., 2008a; Tan et al., 2010b), but are further inhibited when Dz13 is delivered with a nanoparticle combined with intraperitoneally administered doxorubicin (Dox) solution (clinically used against bone tumors), highlighting the usefulness of such combination therapy for tumor death (Dass et al., 2008d).
However, one problem encountered was a typical toxicity to both the heart and skin when free Dox is administered systemically at even these relatively low doses to mice (Ta et al., 2009a,b,c; Tan et al., 2010a), mimicking the human response to this cytotoxic agent. Thus, a method for limiting Dox activity to the lesion site and avoiding normal tissue exposure was deemed necessary. To this end, a drug delivery system (DDS) for Dox was developed. The nanoparticle DDS for Dz13 was developed to overcome reliance on commercial transfection reagents, which have been used for more than a decade (DeCruz et al., 1996; Fahmy et al., 2003; Mitchell et al., 2004). These commercial reagents are not recommended for in vivo use mainly because of their cytotoxicity, in general, against mammalian cells (Dass and Burton, 1999; Dass et al., 2002). One other DDS commonly used for oligonucleotide administration in vivo is the commercial osmotic pump (Alzet®), which can deliver such potentially therapeutic agents such as antisense molecules (Walker et al., 1998, 2002), but when used to administer Dz13, slight hepatotoxicity in mice is noted (Dass and Choong, 2010).
Interestingly, E2F1 (a tumor suppressor) is in fact upregulated in melanoma cells treated with Dox (Hao et al., 2009), as is activation of caspase-2 by this anthracycline in cervical cancer (Tinel and Tschopp, 2004) and leukemic cells (Panaretakis et al., 2005). However, in our hands, for breast cancer and prostate cancer cell lines, free (nonencapsulated) Dox is not active unless micromolar concentrations are used in vitro, whereas for most OS cells, the EC50 is low at ∼50 nM (unpublished data). In contrast, regardless of cancer cell type, Dz13 has an EC50 almost consistently at 400 nM, and this is increased when an encapsulated form of the oligonucleotide is used for treating cells to 250 nM. The former finding has been the case for 14 cancer cell lines (OS, LS, prostate cancer, breast cancer, squamous cell carcinoma, melanoma) that have been tested (Zhang et al., 2004, 2006; Dass and Choong, 2008) and attests to the latent potential of Dz13 as a potent anticancer agent.
Other Agents That Activate Caspase-2
Apart from Dz13, there are other inducers of caspase-2 activation (summarized in Table 2). However, these have only been tested in a very limited number of cancer cell types, which is why we present Dz13 as a molecule that could become important to understanding the role of caspase-2 in cell biology. Not surprisingly, some of these agents are being used clinically to alleviate the burden of disease in patients suffering from a variety of cancers, although the exact mechanism(s) at play are largely unknown. Amongst these, docetaxel is one compound that has been used most frequently for caspase-2 activation.
The initial study linking docetaxel with activation of caspase-2 was performed by Mhaidat et al. (2007a). In their study, apoptotic cell death could be inhibited by overexpression of Bcl-2, even though docetaxel-induced changes in Bax were not inhibited by such overexpression. Caspase-2 activation seemed to be the initiating event of Bax expression, because inhibition of caspase-2 inhibited changes in Bax and mitochondrial membrane potential. Activation of caspase-8 and then Bid seemed to be a late event, although docetaxel was able to induce apoptosis in cells deficient in caspase-8 and Bid. p53 did not seem to be involved as a p53−/− cell line was sensitive to docetaxel and an inhibitor of p53 did not inhibit apoptosis.
In a second study, docetaxel treatment caused activation of both c-Jun N-terminal kinase (JNK) and extracelluar signal-regulated kinases (ERK1/2) but not p38 mitogen-activated protein kinase or Akt kinases (Mhaidat et al., 2007b). Apoptosis was dependent on activation of caspase-2 and downstream changes in Bax and Bak by this caspase, leading to subsequent mitochondrial release of apoptosis-inducing factor and cytochrome c.
In a recent study, docetaxel-loaded [1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy(polyethylene glycol)]2000-stabilized oleic acid-coated hydroxyapatite nanoparticles (Dtxl-NPs) were more cytotoxic against prostate cancer cell lines compared with docetaxel in vitro (Luo et al., 2010). This was linked to increased docetaxel-induced apoptosis in these cells. Prostate cancer cells treated with Dtxl-NPs exhibited significant arrest in the G2-M phase but a higher sub-G0/G1 population when compared with Dtxl. The enhanced apoptosis induced by Dtxl-NPs in the cells was associated with the activation of caspase-2. The study demonstrated an early activation of caspase-2 in Dtxl-NPs–induced apoptosis in cells, which differed from Dtxl-induced apoptosis. When caspase-2 activation was inhibited by small interfering RNA (siRNA) knockdown, a significant inhibition of Dtxl-NPs–induced disruption of MMP and Dtxl-NPs–induced apoptosis was noted.
Dox is an anthracycline anticancer drug molecule that is used against a variety of cancers clinically. A recent study illuminates how this molecule performs its anticancer activities—activation of the initiator caspases-2 and −8 and then of -3 (Fabbri et al., 2011). In a prostate cancer cell line, anthracyclines, in particular a nonpegylated liposomal Dox, were highly concentrated in the Golgi apparatus. The authors hypothesized that the Golgi apparatus, probably acting as a stress sensor, intensified the conventional apoptotic mechanism induced by Dox. They also proposed that apart from the nucleus and the mitochondria, the Golgi apparatus could become an additional new target for anticancer therapy.
The anticancer agent 2-chloro-2′-deoxy-adenosine (Cladribine) and its derivative 2-chloro-adenosine have been shown to induce apoptosis of human astrocytoma cells (Ceruti et al., 2000). The exact pathway involved was not known until more recently (Ceruti et al., 2003), wherein both compounds produced a gradual, time-dependent activation of caspase-3. When caspase-3 activation was suppressed with a chemical inhibitor, apoptosis induction was also inhibited. The initiator caspases-9 and -8 were only marginally activated at later times of apoptosis induction. Intriguingly, neither of the adenosine analogs had any effect on mitochondrial membrane potential, which helped rule out a role for intrinsic apoptotic pathway involving caspase-9 and cytochrome c. As confirmation, no cytosolic accumulation of cytochrome c was detected with either of the adenosine analogs. Interestingly, at early time points after addition of either of the adenosine analogs and preceding the activation of caspase-3, caspase-2 activity was markedly increased. Caspase-2 inhibition with N-benzyloxycarbonyl-Val-Asp-Val-Ala-Asp-fmk significantly reduced both adenosine analog-induced caspase-2 activation and the associated apoptosis. The authors concluded that the adenosine analogs induced apoptosis in these cells by activating an atypical apoptotic cascade involving caspase-2 as the initiator caspase and an effector caspase (caspase-3), although a direct link between the two caspases was not demonstrated.
PRIMA-1MET, a mutant p53-targeting compound, activates caspase-2 in a human lung cancer cell (Shen et al., 2008). As mentioned earlier, it has been known for years that p53 is essential for caspase-2 activation (Vakifahmetoglu et al., 2006). Moreover, the early activation of caspase-2 post-PRIMA-1MET treatment supports the initiator caspase nature of this enzyme. This assumption was further strengthened by recent data demonstrating that both caspase-2 inhibition and siRNA knockdown blocked PRIMA-1MET-induced cytochrome c release and downstream caspase activation (Shen et al., 2008). PRIMA-1MET treatment caused increased levels of the small PIDD-CC fragment in treated lung cancer cells but not in the parental p53−/− cells. The formation of this fragment is strongly correlated with the apoptotic response of cells and is preceded by activation of caspase-2 (Tinel and Tschopp, 2004).
Future Directions
In addition to caspase-2, in some cases (Dass et al., 2008b, 2008e), caspase-8 is also involved in Dz13-mediated apoptotic death of tumor cells. Further, it has been demonstrated that the piddosomal components of PIDD, RAIDD, and DNA-PKcs are all not necessary for caspase-2 activation by Dz13 (Dass and Choong, 2010). Dz13 can then be used to examine whether the death-inducing signaling complex (DISC), which is required for caspase-8 activation, is responsible for caspase-2 activation in tumor cells (Fig. 2), as has been shown in cells with p53 (Olsson et al., 2009). Dz13 may be acting through the death receptor pathway of apoptosis, which involves extracellular ligands interacting with members of the TNF receptor family to recruit the adaptor molecule Fas (CD95)-associated protein with death domain (FADD). These findings will shed light on the interactions between caspase-2 and caspase-8 via FADD. This will be an important basic biology question to answer, especially whether it occurs in the absence of the tumor suppressor p53 protein, which is thought to induce this interaction (Olsson et al., 2009). The latter has not been reported to date and would lend valuable information to this field of research. The study will also elucidate the mechanisms behind Dz13's proapoptotic activity as it pertains to caspase-2 and -8 and FADD.
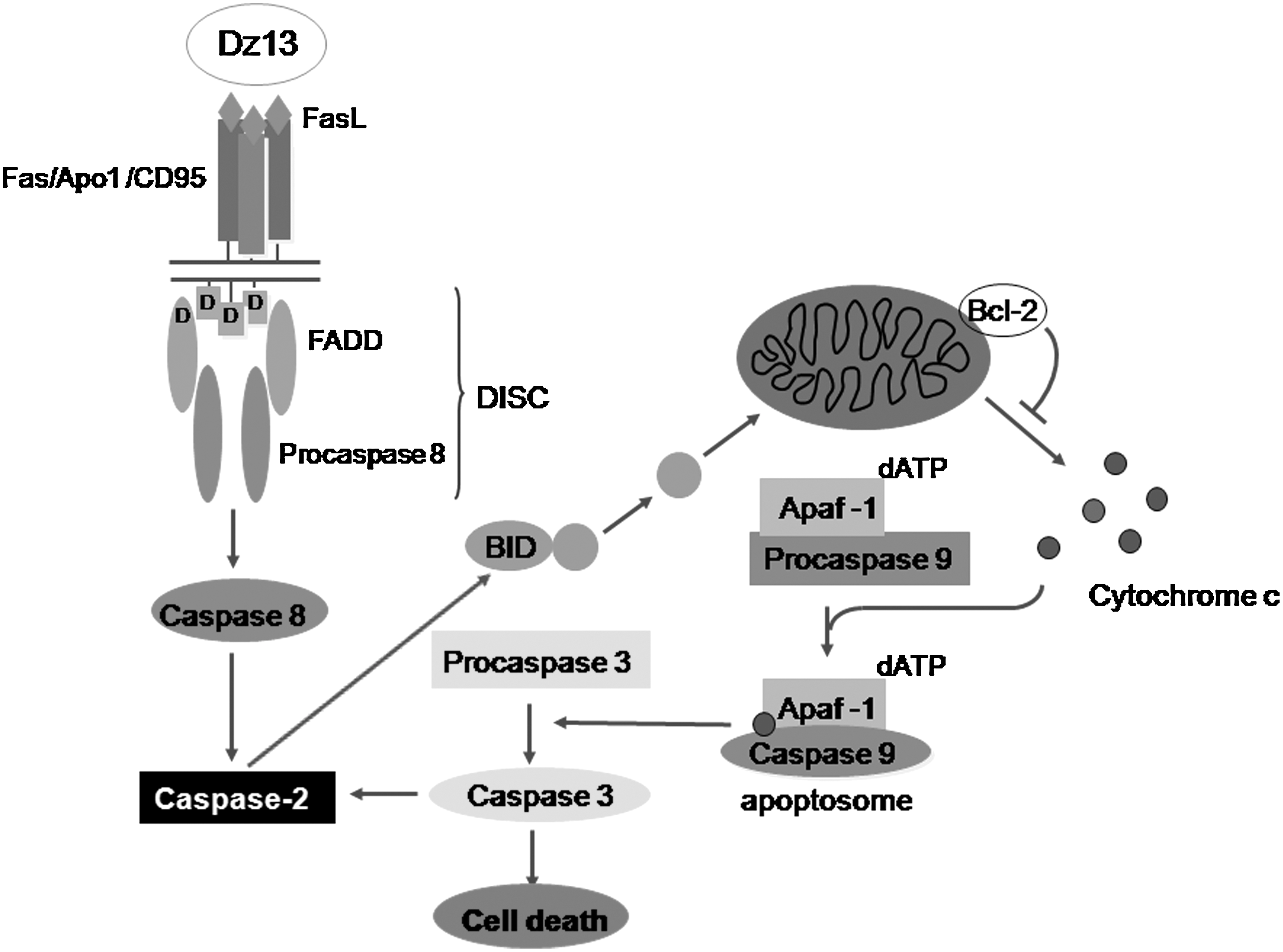
Dz13 induces FADD/caspase-2 apoptotic pathway in mammalian cells. Caspase-2–dependent death can be initiated in membrane-based activation platforms (DISC). Subsequent mitochondrial membrane permeabilization, involving BH3-interacting domain (Bid) processing, then ensues. Caspase-2 is also reported to contribute to a mitochondrial amplification loop downstream of caspase-3 activation. Adapted from the works of Hengartner (2000), Green and Kroemer (1998), and Krumschnabel et al. (2009). Dz13, DNAzyme 13; DISC, death-inducing signaling complex.
Targeted therapy using biological agents in combination with conventional chemotherapy has the potential to increase the efficacy of treatment while reducing morbidity. As caspase-2 is central to Dz13 proapoptotic activity, formalin-fixed, paraffin-embedded clinical specimens of OS were analyzed for caspase-2 protein immunohistochemically (Dass and Choong, 2010). Importantly, all the tumors expressed caspase-2, with expression being sporadic and localized to various aspects of the tumors, in areas exhibiting growth spurts and more mature sites such as osteoid tissue. Such widespread distribution of caspase-2 in tumor tissue will assist in a wider cell death response with an agent capable of activating caspase-2, such as Dz13.
Summary
As methods for detection become more sensitive and widespread, the incidence of cancer has been noted to be on the rise globally. Evasion of apoptosis, a type of cell death, is a hallmark of tumor growth and progression. Central to apoptosis are a series of cysteine protease proteins known as caspases, responsible for widespread destruction in cells programmed to die. Caspase-2, an enigmatic caspase increasingly being associated with certain types of cell death in cells exposed to cytotoxic agents, is one that little is known of compared with its counterpart caspases. Part of the reason for this enigma has been lack of a suitable stimulus for this caspase, and with the discovery of Dz13, a potent inducer, much more can now be elucidated. For instance, one thing that could be unraveled is whether caspase-8 and FADD are indeed involved in caspase-2 activation as part of the DISC. Finally, a better understanding of caspase-2 biology may lead to more effective ways of treating tumors. Indeed, most tumor cells tested to date are inhibited by Dz13, a potent inducer of caspase-2 activation.
Footnotes
Acknowledgment
The authors thank the Victorian University Research and Development Grant Scheme (VU-RDGS) for financial support.
Disclosure Statement
The authors declare that there is no conflict of interest in relation to writing this manuscript.