
Abstract
Head and neck cancers (head and neck squamous cell carcinomas [HNSCC]) are a heterogeneous group of neoplasms with varying presenting symptoms, treatment, and expected outcome. There is a need to find an effective way of its treatment at the molecular level. Thus, we should identify the mechanism of cancer cell response to damaging agents' activity, especially at DNA level. Our major goal was to evaluate the efficacy of DNA double strand breaks (DSBs) repair in HTB-43 and SCC-25 cancer cell lines as well as lymphocytes taken from HNSCC patients and healthy donors. The DNA repair efficiency was measured by neutral comet assay as well as extrachromosomal assay for DNA DSBs repair (TAK assay). We determined the levels of two main pathways of DNA DSBs—nonhomologous end joining (NHEJ) and homologous recombination repair (HRR). Neutral comet assay was used for evaluation of DNA DSBs repair after treatment with genotoxic agents. DNA DSBs induced by gamma radiation were repaired slower in lymphocytes from HNSCC patients than in lymphocytes from healthy controls. HTB-43 and SCC-25 cancer cell lines have higher efficacy of NHEJ and HRR than lymphocytes taken from patients as well as control subjects. Our results confirm the necessity of further studies on the mechanisms of DNA DSBs repair to provide insight into the molecular basis of head and neck cancer, which will allow us to improve methods of HNSCC treatment.
Introduction
The clustered nature of energy deposition events generated by ionizing radiation leads to the formation of DNA DSBs and other complex lesions, mainly by generation of free radicals, but also by direct interaction with DNA molecule (Vamvakas et al., 1997; Khanna and Jackson, 2001). Epidemiological, biological, and clinical studies have provided various lines of evidences that free radical-induced oxidative damage of cell membranes, nucleic acids, and proteins might play a causative role in aging and several degenerative diseases, such as cancer, atherosclerosis, cataracts, as well as Alzheimer's disease (Longhese et al., 2006; Halazonetis et al., 2008; Weyand et al., 2009).
Cells of higher eukaryotes display an astonishing capacity to remove DSBs from their genome using two conceptually different repair pathways. The first repair pathway utilizes homology available elsewhere in the genome to restore structural integrity and sequence fidelity in the DNA molecule. This mechanism, called homologous recombination repair (HRR), operates mostly in a error-free manner, is relatively complex and therefore slow, and is optimized to function after DNA replication using the sister chromatid as a source of homology (Li and Heyer, 2008). The second repair pathway is optimized to rejoin DNA DSBs without any homology requirement, in a cell cycle independent manner and to restore structural integrity, but not sequence fidelity, in the molecule. This mechanism, termed nonhomologous end joining (NHEJ), is capable of quickly removing DSBs from the genome (Weterings and Chen, 2008).
It is still not known what mechanism is responsible for successful radiotherapy at the molecular level. Therefore, the aim of our research was to evaluate the level of DNA DSBs repair in cells treated with γ-radiation, generating free radicals and causing other types of damages by direct interaction with many types of molecules, H2O2, the source of reactive oxygen species, and those electroporated with external DNA digested with restriction enzymes to introduce DNA DSBs. The goal of this study was to show the differences between the DNA DSBs repair level in cells affected by different cancer treatment agents. DNA integrity is the main factor responsible for overall survival of cancer cells. Therefore, our study also assessed whether introducing DNA double strand damage is sufficient to kill the cancer cell, or if the efficiency of cancer treatment is more complex and involves damaging other cellular components. Our study supports emphasizing the role of the proteome and other cell structures in maintaining the cellular stability.
Materials and Methods
Study subjects
Examined material comprised lymphocytes isolated from peripheral blood as well as head and neck cancer squamous cell lines—HTB-43 and SCC-25. The group of patients enrolled in our study consisted of 8 women and 10 men (mean age 61±9 years). All subjects were found to have HNSCC (most of them with larynx cancer); five of them had metastases to lips, tongue, lymph nodes, and salivary glands. Patients did not receive any chemotherapy or radiotherapy for their primary disease before blood sample collection. Twenty healthy subjects (9 women and 11 men at the same age as patients) were also involved in the study and treated as a control group. Controls were selected based on family history, to exclude familiar predisposition to cancer development. Before examination, patients and control subjects did not receive other drugs such as antibiotics or steroids. All subjects involved into the study were unrelated Caucasians and lived in the Lodz district, Poland. Study material was obtained in Department of Otolaryngology and Oncology, Medical University of Lodz, Poland. The study was approved by the Local Ethic Committee and a written consent was obtained from each subject before enrolling into the study. The HTB-43 larynx cancer and SCC-25 tongue cancer cell lines (commercially available; ATCC, Manassas, VA) were also used in our trial.
Cell treatment
The HTB-43 larynx cancer cell line was cultured in EMEM (ATCC) (mediaalso contained 10% fetal bovine serum, penicillin and streptomycin). The SCC-25 tongue cancer cell line was cultured in DMEM/F-12 (ATCC) (media also contained 10% fetal bovine serum, hydrocortisone, penicillin and streptomycin; Sigma-Aldrich, St. Louis, MO). Both cell lines were kept at 37°C in an atmosphere containing 5% CO2. Lymphocytes were isolated from peripheral blood of HNSCC patients and healthy donors by centrifugation in a gradient of Histopaque-1077 (Sigma-Aldrich). After isolation lymphocytes were washed with phosphate-buffered saline (PBS) and suspended in RPMI-1640 growth medium (ATCC) for further treatments. γ-irradiation was carried out using a 60 Co source (Technical University of Lodz, Lodz, Poland) at a 0.0266 Gy/s dose rate. The cells were kept at room temperature during irradiation with 5–30 Gy. Hydrogen peroxide (Sigma-Aldrich) treatment was prepared in final concentrations of 0–50 μM. The initial viability of cells in each experiment was about 90%, as measured by the trypan blue staining exclusion method.
MTT assay
After irradiation or incubation with hydrogen peroxide, the resistance of cells was evaluated by the MTT assay. H2O2 was added to the cell suspensions (1.5×10/mL of growth medium) in concentration of range 0–50 μM. Investigated cells were also irradiated with 0–30 Gy at concentration of 1.5×106 cells/mL in the growth medium. After treatment, all samples were placed onto 96-well plates in 150 μL of appropriate growth medium and 20 μL of 10 mg/mL MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] was added to each well. After incubation at 37°C for 4 h, the supernatant was removed and 200 μL of 10% sodium dodecyl sulfate and 0.04 M HCl was added to dissolve the formazan salt. One hour later, the difference of absorption intensity (OD650–OD570 nm) was measured by an ELISA microplate reader (Bio-Rad, Hercules, CA). The resistance was presented as a percentage of viable cells in suspension culture exposed to hydrogen peroxide or γ-radiation in comparison to the untreated control cells. All chemicals used were from Sigma-Aldrich.
Comet assay
To detect the level of DNA damage and repair efficiency, the single cell gel electrophoresis assay was performed. The assay was done under neutral conditions according to the procedure of Singh et al. (1988) with modifications by Klaude et al. (1996).
To induce the DNA damage, lymphocytes as well as HTB-43 and SCC-25 cells (1–2×105/mL) were treated with hydrogen peroxide in concentrations of 0, 2.5, 5, 10, 15, and 25 μM for 10 min at 4°C, washed, and resuspended in a fresh, drug-free growth medium. In a separate experiment, cell suspensions were irradiated at doses of 0, 5, 15, 25, 35, and 50 Gy. To evaluate the kinetics of DNA repair, after the procedure described above the cells suspensions were allowed to repair for 30, 60, 120, and 240 min or were harvested immediately (time 0=no repair incubation) then placed on ice. The cells were suspended in 0.75% low-melting agarose dissolved in PBS and layered onto microscope slides (Superior, Germany) previously covered with 0.5% normal melting point agarose (Sigma-Aldrich). Cells were lysed at 4°C for 1 h in a buffer consisting of 2.5 M NaCl, 100 mM EDTA, 1% Triton X-100, and 10 mM tris(hydroxymethyl)aminomethane, pH 10 (Sigma-Aldrich). Next, the DNA was unwound and uncoiled under normal conditions (pH=8) for 40 min in the electrophoresis buffer consisting of 300 mM NaOH and 1 mM EDTA (Sigma-Aldrich). Electrophoresis was carried out at 4°C for 30 min at an electric field strength equal to 0.73 V/cm (30 mA),then samples were neutralized with 0.4 M tris(hydroxymethyl)aminomethane, pH 7.5. Next, cells were stained with DAPI solution (2 μg/mL), covered with a cover glass and incubated at 4°C for 45 min. All of the steps described above were performed under dimmed light or in the dark in order to prevent additional DNA damage. The slides were examined at 200× magnification with an Eclipse fluorescence microscope (Nikon, Tokyo, Japan) attached to a COHU 4910 video camera (Cohu, Inc., San Diego, CA) equipped with a UV filter block. One hundred images were randomly selected from each sample and the comet tail moment (a product of fraction of DNA in tail and tail length) was measured. Two parallel tests with aliquots of the same sample were performed for a total of 200 cells and the mean comet tail moment was calculated. Each experiment was repeated three times. The comet tail moment is positively correlated with the level of DNA breakage in a cell. A mean value of tail moment in particular sample was taken as an indicator of DNA damage in the sample.
TAK assay
To delineate the nature of the DNA DSBs repair mechanism, a three-plasmid TAK (Tetracycline, Ampicillin, and Kanamycin) assay based on bacterial transformation was utilized (Liang and Jasin, 1999). Plasmid pCGJKm with kanamycin resistance (KmR) gene was introduced undigested into mammalian cells to serve as a transfection control. Plasmid pBR322 containing ampicillin- (AmpR) and tetracycline-resistance (TetR) regions was used to measure the levels of DNA HRR and NHEJ. The AmpR gene was digested with EcoRV and NheI, and the TetR gene with NruI and XmaIII restriction enzymes (New England Biolabs, Ipswich, MA). Fragments of digested plasmids were separated on agarose gel during electrophoresis and then were recovered by using a Millipore elution kit (Millipore QIAquick Gel Extraction Kit; QIAGEN, Valencia, CA). The plasmid backbones of pBR322Amp and pBR322Tet are each capable of being recircularized in mammalian cells to measure DNA end joining. When recovered from mammalian cells and reintroduced into bacteria, the recircularized plasmids confer AmpR, which serves as the measure of DNA end joining. In a separate experiment, the pBR322Tet and pBR322Amp plasmids were also used to measure DSB-promoted HR in mammalian cells. Plasmid pBR322Tet contains the 5′ end, and pBR322Amp contains the 3′ end of the TetR gene with 552 bp of homology between them. The plasmid backbones are also homologous. When the two plasmids recombine within the tat homology region, the TetR gene is recovered, and the recombined plasmid confers TetR. HR between two circular plasmids is inefficient in mammalian cells but is greatly stimulated when a DSB is introduced next to the homology region (Liang and Jasin, 1996). Thus, pBR322Tet linearized with NruI and XmaIII was then digested with SspI (New England Biolabs) to release the homologous TetR fragment. The two linear plasmids and the small fragment of the TetR gene were all separated on agarose gel during electrophoresis to detect HRR. All the plasmid substrates were electrotransfected into HTB-43 and SCC-25 cells, lymphocytes taken from patients with head and neck cancer as well as lymphocytes taken from healthy donors using Electrotransfomation Module Gene Pulser Xcell (Bio-Rad) at 1000 μF/155 V at 0.2 cm cuvette (Bio-Rad). Then, DNA from transfected cells was recovered after 4 h of repair incubation by phenol/chloroform extraction and transformed into Escherichia coli DH5α (Invitrogen, San Diego, CA). Colonies that grew with selective antibiotics were scored, and then the plates were washed with 2 mL PBS to analyze DNA repair products. The TAK assay estimates DNA DSBs repair, since number of KmR colonies measured transfection efficiency, AmpR indicated DNA end joining, and TetR indicated HR. A control experiment to verify if HR occurs indeed in the mammalian cells and not in E. coli was made by using the separate incubation of the linear substrates in mammalian cells and combining the all DNA isolated just before bacteria transformation. No colonies resistant to tetracycline were observed (data not shown).
Statistical analysis
The data from MTT and TAK assays were expressed as mean±standard deviation from experiments performed on each subject (patients, healthy subjects) as well as cell lines. The values from the comet assay were expressed as mean±standard error of the mean. If no significant differences between variations were found, as assessed by Snedecor-Fisher test, the differences between means were evaluated by t-test. Otherwise, the Cochran-Cox test was used. The data were analyzed using STATISTICA 6.0 (StatSoft, Tulsa, OK) statistical package. One hundred cells were scored to estimate proliferation ability of the cell lines.
Results
Cells viability
Figure 1 shows the mean percentage of the viability of lymphocytes from healthy donors used as a control, lymphocytes from HNSCC patients as well as HTB-43 and SCC-25 cells measured by the MTT test after 3 days of incubation with an increasing concentration of hydrogen peroxide from 2.5 to 50 μM or irradiated with an increasing dose of γ-radiation from 5 to 50 Gy. HNSCC cells were more sensitive to genotoxic treatment than control cells. With the highest dose of hydrogen peroxide, 50 μM, the viability of the control cells decreased to 22% and for lymphocytes from HNSCC patients the average was 4%. At a dose of 25 μM, we observed rapid decrease of HTB-43 cell viability. The most insusceptible to H2O2 treatment were lymphocytes taken from healthy subjects. The most susceptible to γ-irradiation were SCC-25 cells and HNSCC patients' lymphocytes (40% and 54.5%, respectively, at a dose of 50 Gy), whereas viability of lymphocytes taken from healthy subjects was about 80%. At lower doses of irradiation (up to 20 Gy) HTB-43 cells and lymphocytes taken from patients with cancer presented higher viability than control lymphocytes. Statistically significant difference (p<0.001) was observed between all types of investigated cells in comparison with control lymphocytes.

The viability of lymphocytes from healthy donors (—•—), lymphocytes from HNSCC patients (—○—), HTB-43 (—▾—) and SCC-25 cells (—▵—) measured by MTT assay after treatment with H2O2 (upper panel) or γ-radiation (lower panel). Error bars indicate standard deviation. HNSCC, head and neck squamous cell carcinoma.
DNA damage
Figure 2 shows DNA damage measured by comet assay as a mean percentage of DNA present in the comet tail (Tail DNA%) of lymphocytes form healthy donors (Fig. 2A), lymphocytes from HNSCC patients (Fig. 2B), HTB-43 cells (Fig. 2C), and SCC-25 cells (Fig. 2D) after hydrogen peroxide treatment with increasing concentrations from 0 to 25 μM. In every type of investigated cell, we observed statistically significant increase of DNA damage (p<0.001) in a dose-dependent manner. The most susceptible to hydrogen peroxide treatment were lymphocytes taken from patients with head and neck cancer. In this type of cell we observed the rapid increase of DNA damage (Tail DNA%) at a concentration of 5 μM. At the higher doses (from 10 to 25 μM) in lymphocytes taken from HNSCC patients we observed the highest level of damage among all investigated cell types. The lowest level of DNA damage after hydrogen peroxide treatment was observed in the SCC-25 cell line—Tail DNA% did not exceed 17% at the highest concentration of H2O2.

Mean percentage of DNA damage after H2O2 treatment. The level of DNA breakdown was measured in lymphocytes form healthy donors
In separate experiments, cells were treated with γ-radiation. The effects of irradiation are presented in Figure 3. Evaluated cells showed diverse susceptibility to gamma radiation. At a dose of 5 Gy, the highest level of DNA breaks (8.97%) was observed in HTB-43 cells, but at 15 and 25 Gy, lymphocytes taken from patients with HNSCC had higher susceptibility to irradiation (18.62% and 21.30%, respectively). Contrary to results obtained after hydrogen peroxide treatment, among the irradiated cells the highest level of DNA damage (32% for 35 Gy and 44.4% for 50 Gy) was observed in lymphocytes from healthy donors. The most resist to γ-radiation at all doses was the SCC-25 cell line where the lowest DNA damage was observed among all types of evaluated cells.
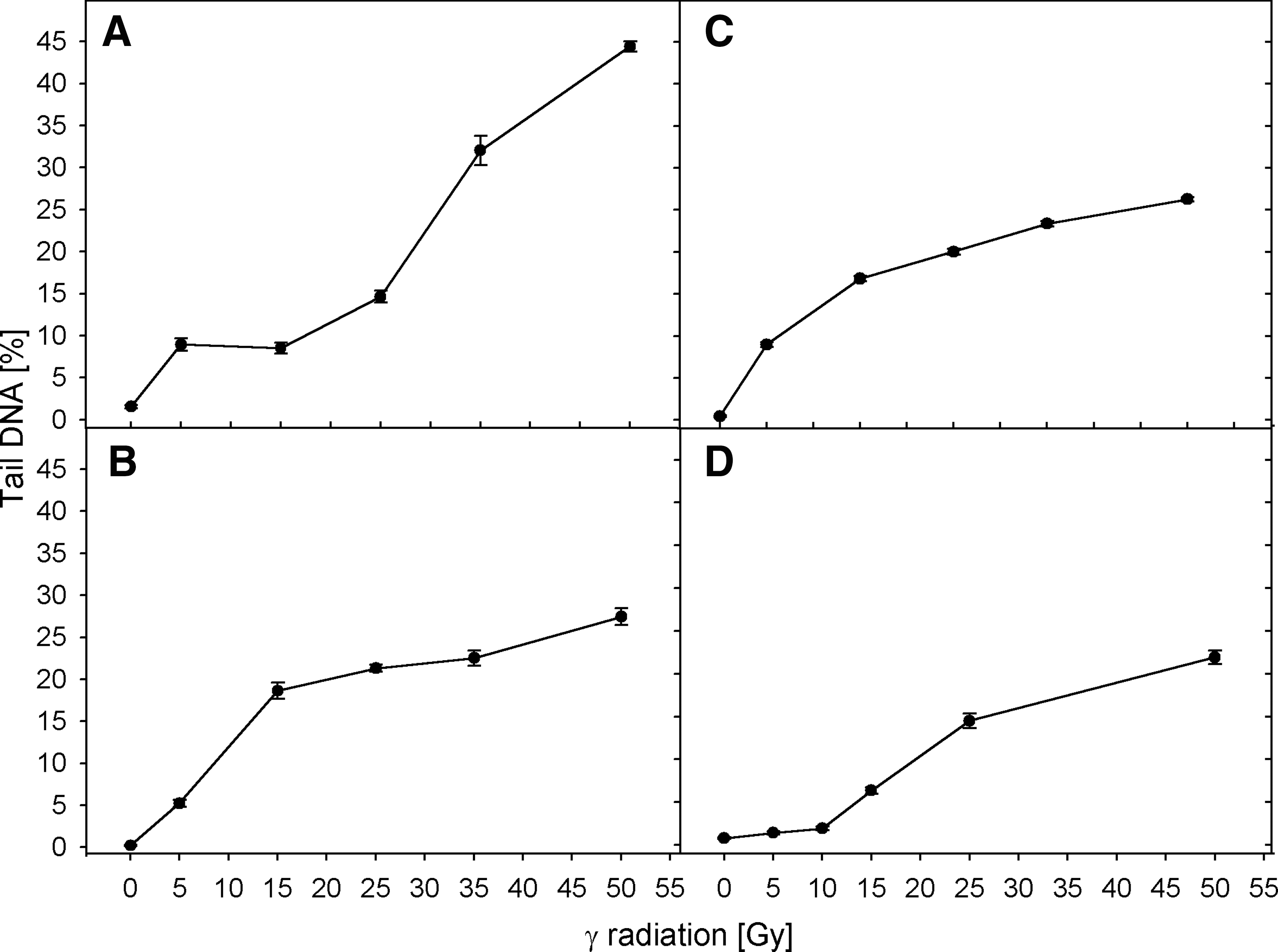
The mean percentage of DNA damage after γ-irradiation. The level of DNA breakdown was measured in lymphocytes form healthy donors
DNA repair
Figures 4 and 5 present the kinetics of rejoining of DNA double-strand breaks after hydrogen peroxide treatment and irradiation, respectively. The DNA damage was measured as a mean percentage of DNA in the comet tail (Tail DNA%). In all cases, the level of DNA damage in the control cells was constant, which indicates that the preparation procedure did not induce significant DNA damage. In all investigated cell types, we observed the statistically significant decrease of Tail DNA% level in comparison to control (p<0.001).
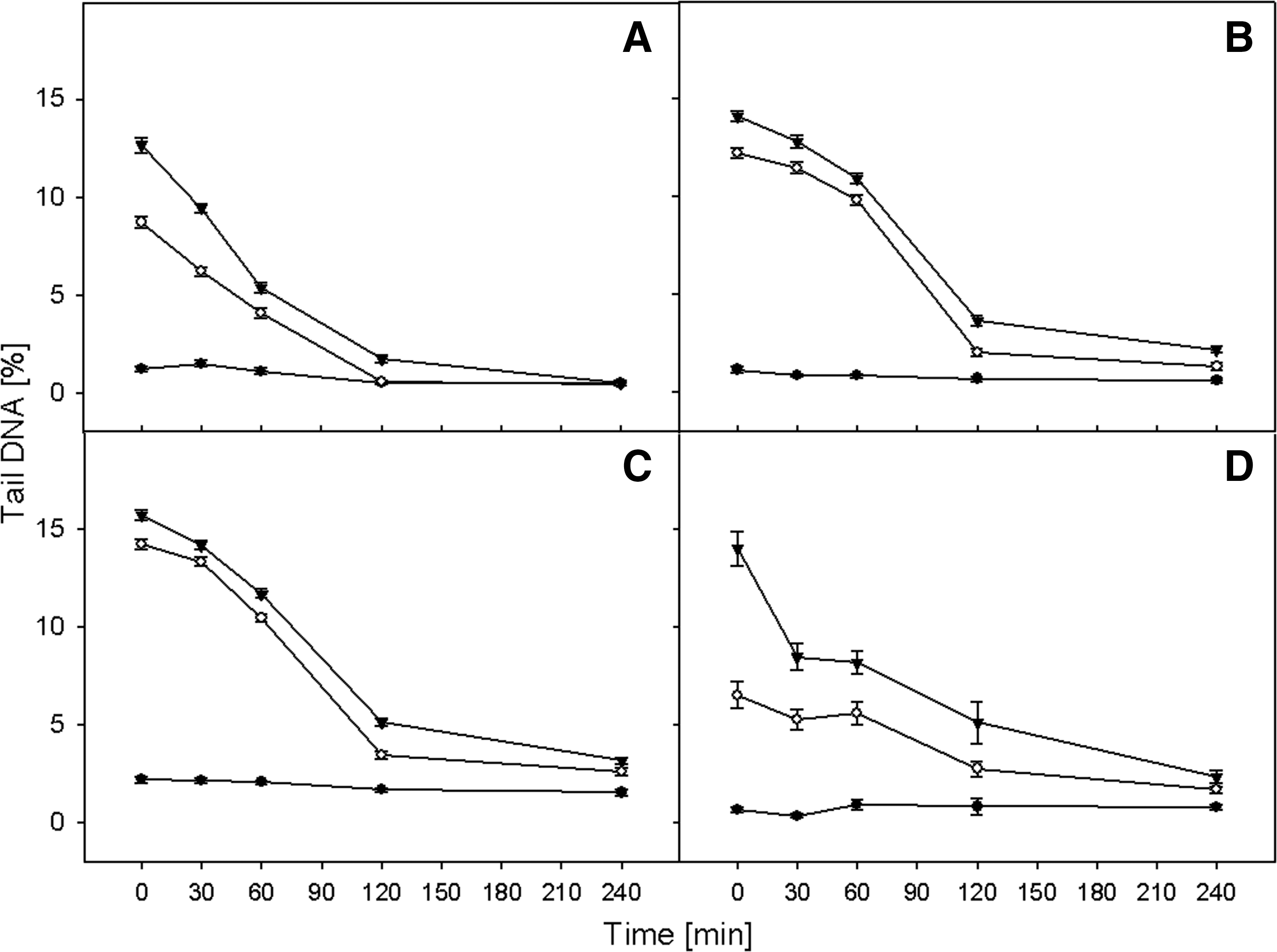
The kinetics of DNA double strand breaks repair after H2O2 treatment. The course of recovery was measured in lymphocytes form healthy donors
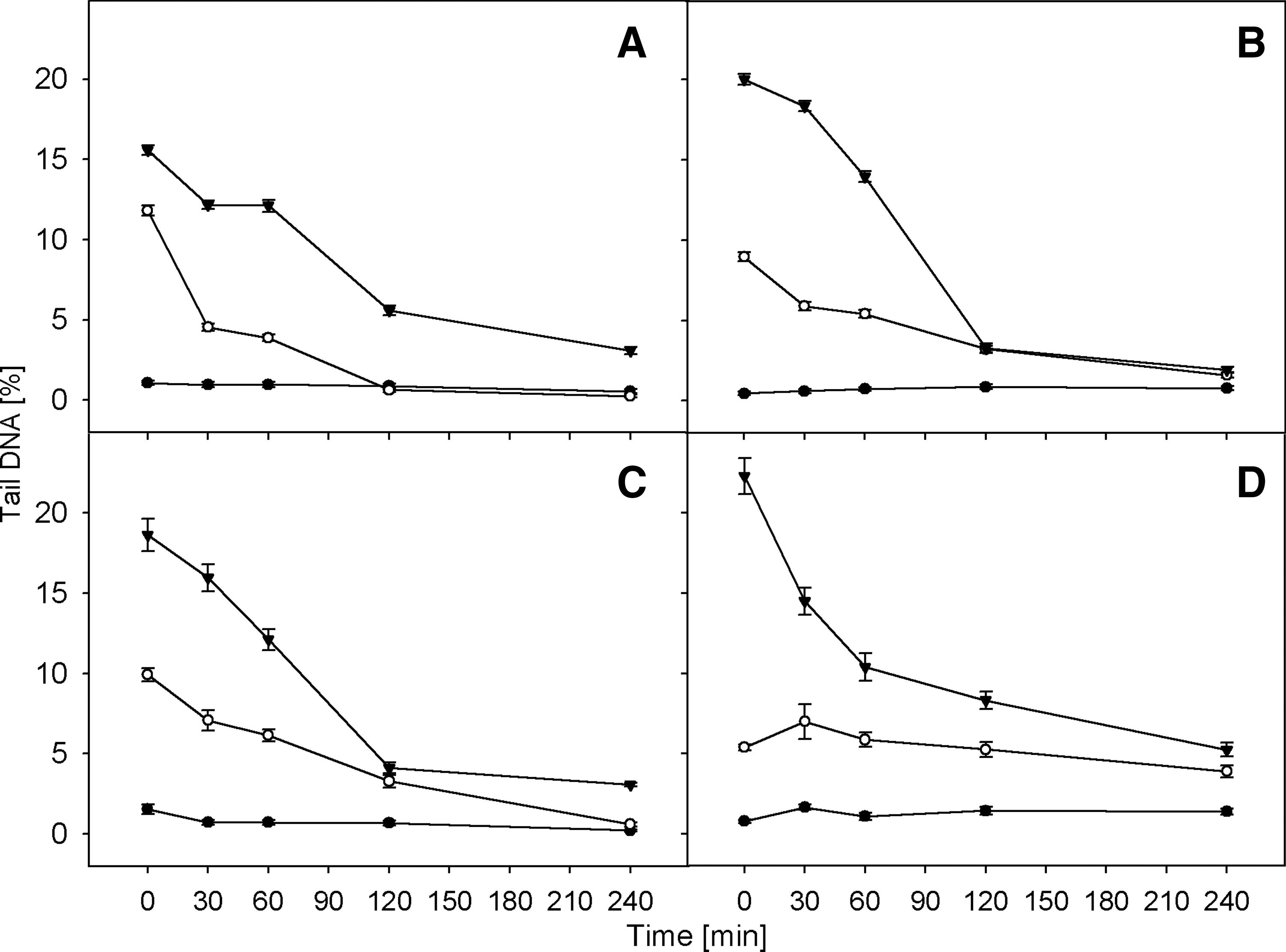
The kinetics of DNA double strand breaks repair after γ-irradiation. The course of recovery was measured in lymphocytes form healthy donors
Cells were preincubated with two doses of H2O2 (5 and 10 μM) and compared to unexposed cells. Almost all types of investigated cells incubated with the lower concentration of hydrogen peroxide after 240 min of repair incubation had the same level of DNA damage as control cells (Figure 4). The only exception was the SCC-25 cell line. At the higher dose (10 μM), only the DNA in lymphocytes taken from patients with head and neck cancer were completely repaired after 4 h. The lowest repair efficacy was seenin SCC-25 cells.
In a separate experiment, cells were treated with 5 or 25 Gy gamma radiation followed by repair incubation (Fig. 4). Similar to results obtained after preincubation with hydrogen peroxide, the most rapid DNA repair in cells treated with γ radiation was observed up to 120 min. Later the kinetics of DNA repair dramatically slowed down.
After irradiation, only the lymphocytes taken from healthy subjects were able to completely recover—at doses of 5 Gy after 120 min and 25 Gy after 240 min. After 4 h of repair incubation, all types of investigated cells were able to almost complete repair—observed DNA damage level was lower than 5%.
TAK assay
The obtained results suggest that extrachromosomal plasmids were repaired in all types of investigated cells. All bacterial colonies were harvested. Plasmids isolated from those cells were digested by restriction enzymes in order to retrieve the linear form. Electrophoresis was performed to check the size of recovered plasmids (Figure 6).
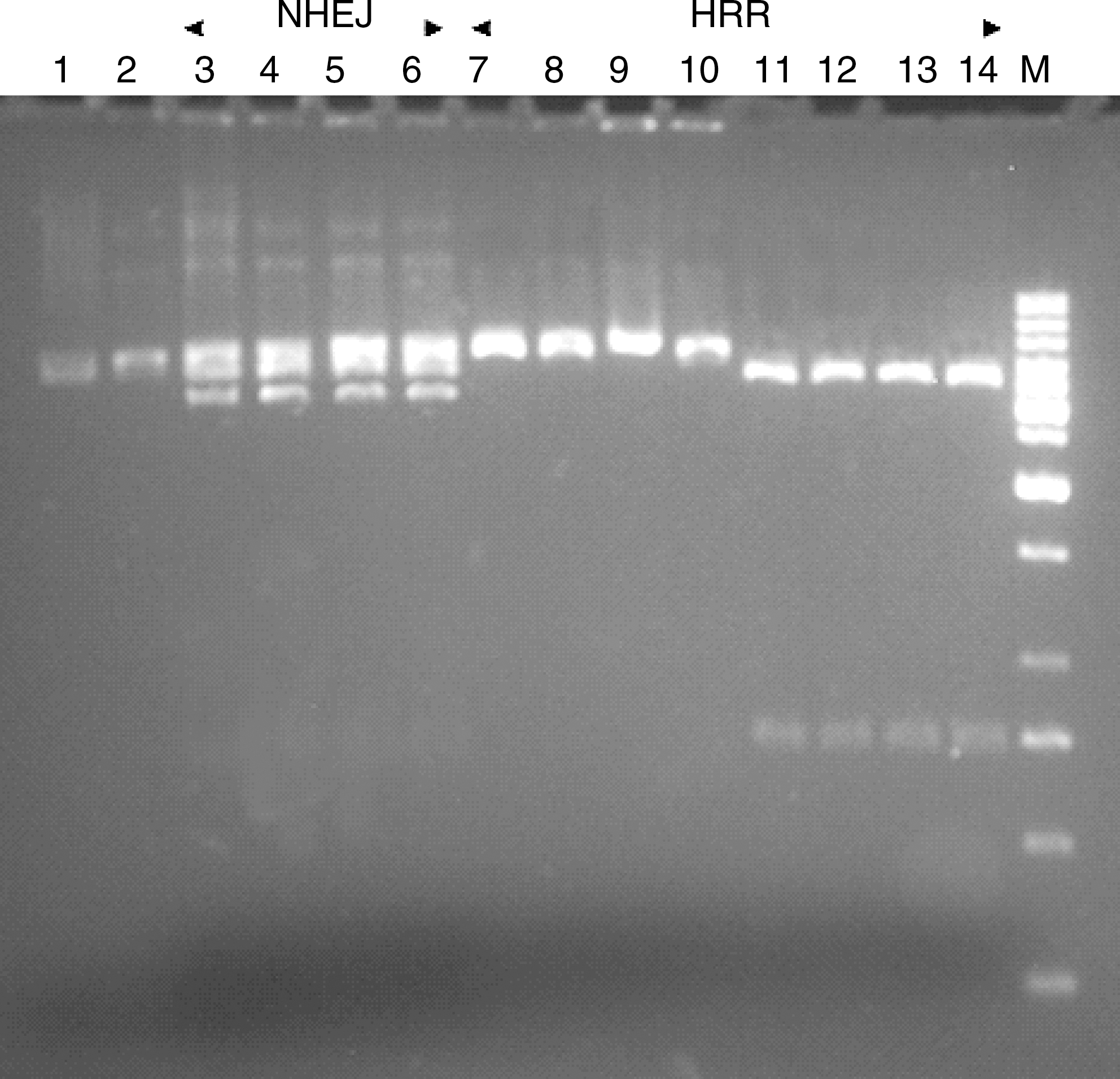
The analysis of repaired plasmid substrates isolated from colonies which grew in medium with Km, Amp, and Tet. Substrates were separated on a 1.5% agarose gel: Lane 1, undigested pBR322 control plasmid; 2, pBR322 control plasmid digested with EcoRI; 3–6, plasmids repaired in lymphocytes from healthy donors, lymphocytes from HNSCC patients, HTB-43 cells and SCC-25 cells, respectively, and isolated from Amp-resistant colonies digested with EcoRI; 7–10, plasmids repaired in lymphocytes from healthy donors, lymphocytes from HNSCC patients, HTB-43 cells and SCC-25 cells, respectively, and isolated from Tet-resistant colonies digested with EcoRI; 11–14, plasmids repaired in lymphocytes from healthy donors, lymphocytes from HNSCC patients, HTB-43 cells and SCC-25 cells, respectively, and isolated from Tet-resistant colonies digested with XmaIII and EcoRV; M, DNA ladder 10 kbp. Km, kanamycin; Amp, ampicillin; Tet, tetracycline; NHEJ, nonhomologous end joining; HRR, homologous recombination repair.
The restriction analysis of DNA recovered from TetR colonies confirmed that the full tetracycline gene is present in HRR products. The analysis of DNA recovered from AmpR colonies revealed the presence of different size products resulting from DNA-ends processing of the linear substrates by NHEJ (Slupianek et al., 2005). Figure 7 displays the efficacy of DNA DSBs repair measured by number of colonies per μg of DNA introduced into bacterial cells. The transfection control pCGJKm was used to calculate the repair efficiency.
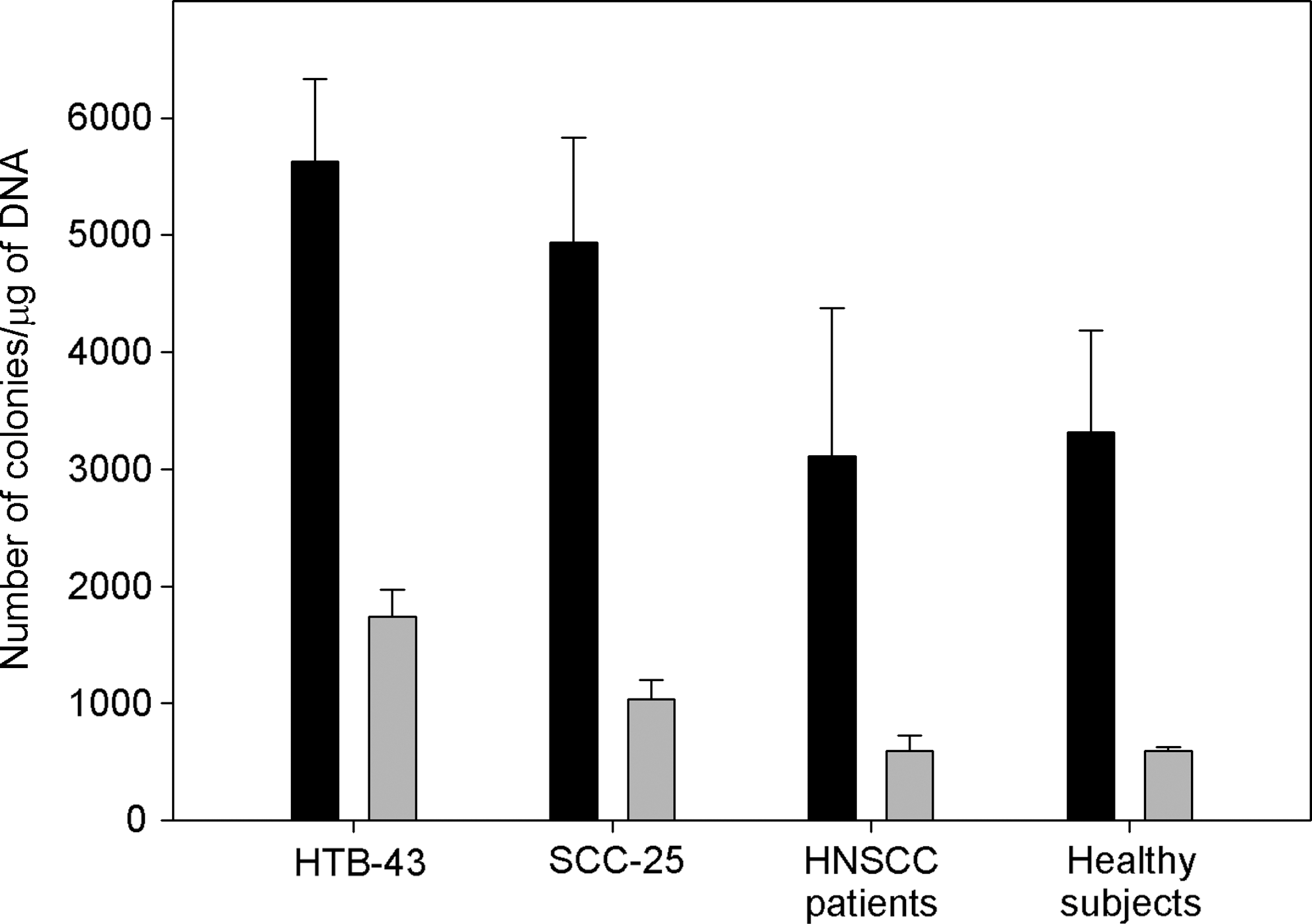
Double strand break repair of extrachromosomal substrates. Figure presents the efficacy of plasmid repair in HTB-43 and SCC-25 cell lines as well as lymphocytes from HNSCC patients and lymphocytes from control group. Black bars indicate the level of NHEJ and gray bars indicate HRR. Results represent mean number of colonies per μg of DNA transformed to E. coli DH5α cells in three independent experiments. Error bars indicate SEM.
Obtained results showed considerable predominance of NHEJ over HRR pathway in all types of investigated cells. Results showed that HTB-43 and SCC-25 cells have higher ability to repair DNA DSBs than lymphocytes. Figure 7 shows the number of colonies per μg of DNA introduced into competent bacteria. The efficiency of NHEJ repair pathway was 5628 colonies/μg DNA for HTB-43 and 4936 colonies/μg DNA for the SCC-25 cell line. In lymphocytes, the NHEJ efficacy was 3110 colonies/μg DNA for cells obtained from HNSCC patients and 3313 colonies/μg DNA for cells of healthy donors. The HRR effectiveness was as follows: 1740 colonies/μg DNA for the HTB-43 cell line, 1035 colonies/μg DNA for the SCC-25 cell line, whereas in lymphocytes obtained from both cancer patients and healthy subjects, efficiencies were equal and amounted to 597 colonies/μg DNA. Statistical analysis performed by t-test showed a significant difference between the levels of repair by HR in HTB-43 and SCC-25 (p=0.012) cell lines, but there were no statistically significant differences in the level of NHEJ repair between these cells (p=0.353). In the case of squamous cell carcinoma lines of the larynx, statistically significant differences in the levels of NHEJ and HR were observed in comparison to lymphocytes from subjects with head and neck cancer (p=0.04 for NHEJ and p=0.002 for HRR) as well as lymphocytes of healthy people (p=0.023 and p=0.001, respectively). Differences between the level of NHEJ in SCC 25 cells and lymphocytes are not statistically significant (p=0.111 for lymphocytes from people with HNSCC and p=0.088 for lymphocytes from healthy individuals). The opposite association was found for HRR (p=0.002 and p=0.001, respectively). The t-test also showed no statistically significant differences in the levels of repair efficiency by NHEJ and HRR in lymphocytes of healthy subjects in comparison to lymphocytes from patients with head and neck cancer (p=0.83 for NHEJ and p=0.99 for HRR).
Discussion
DNA DSBs are removed from the genome by NHEJ or HRR. Data show that inefficient DNA repair is one cause of carcinogenesis (Khanna and Jackson, 2001). DNA DSBs are important direct consequences of cellular exposure to ionizing radiation. A variety of evidence suggests that DNA DSBs are the key damage type linked to radiation-induced lethality. In particular, the link between DSB and chromosome breakage, which in turn closely correlates with cell death in some cell types, is strongly supportive of this concept (Longhese et al., 2006). Our initial interest in examining damage and repair in single cells was driven by the goal of understanding the resistance of cells to DNA damage induced by radiation and reactive oxygen species. We examined the level of repair by two main pathways of DNA DSBs repair, NHEJ and HR, and how they vary between examined cells.
Available data describing DNA repair kinetics in lymphocytes of head and neck cancer patients after exposition to γ-radiation indicated higher level of endogenous DNA damage and decreased efficacy of DNA repair in comparison to lymphocytes taken from healthy donors. After 15 min of repair incubation, a higher level of DNA damage and slower DNA recovery than in lymphocytes taken from control subjects was observed (Saha et al., 2008). Palyvoda et al. (2002) also showed decreased repair in HNSCC patients. which might, in part, explain the increased DNA damage.
Those data are supported by results obtained by Essers and colleagues (1997) as well as Thacker (2005). They revealed that disturbances in the HRR pathway are involved in higher radiosensitivity of patients with cancer. Inactivation of the RAD54 gene as well as mutations in the XRCC2 and XRCC3 genes cause higher susceptibility to irradiation. Data also indicate that mutations in NHEJ genes are the causes of impaired DNA DSBs repair and lower resistance to gamma radiation (Rothkamm and Löbrich, 2003). Our results are also comparable with those obtained by Chen et al. (2010). A similar dose-dependent susceptibility to DNA damaging agents, measured by comet assay, was observed in the SCC-4 cancer cell line.
The studies above describe outcomes of reactive oxygen species or radiation influence on whole cell functioning. Those factors affect not only nucleic acids, but also other molecules present in cells. Reactive oxygen species (ROS) and gamma radiation strongly influence cellular metabolism, which may lead to additional damage. The TAK assay showed that the ratio of NHEJ/HRR effectiveness was similar to those obtained previously by Liang and Jasin (1996). HR products were underrepresented relative to end joining products in the TAK assay, since only one repair event is needed to rejoin the plasmid backbones to confer AmpR, whereas two repair events are required to reclose the plasmid to confer TetR. We observed the significant increase of DNA repair efficiency in cancer cells in comparison to lymphocytes. In the HTB-43 cell line, we observed a 1.7-fold higher level of NHEJ and 2.9-fold higher HRR efficacy, and for the SCC-25 cell line these ratios were 1.49 and 1.73, respectively. The trial did not show statistically significant differences between lymphocytes taken from healthy donors and patients with head and neck cancer. We observed some discrepancy between results obtained in the comet and TAK assays. Our results may suggest that the cause of impaired DNA repair in cells is not only the direct influence of radiation or free radicals on DNA strands, but also their effects on the proteins involved in DNA repair or that they cause other types of damage within the cell. This statement is corroborated by results obtained by Pietrowska et al. (2011). They claimed that abundances of several plasma peptides correlate with either the level of DNA breaks induced in lymphocytes irradiated in vitro or the rate of repair of such damage. They found that the rates of escalation of the acute mucosal reaction (AMR) in head and neck cancer correlated with the levels of residual unrepaired DNA breaks in lymphocytes. Moreover, a faster rate of the AMR in vivo was related to a higher level of unrepaired damage in vitro (p<0.031).
While chromosome aberrations may be a result of DNA double strand breaks, it is very interesting to investigate other factors associated with chromosome pathology and cell division. Inhibition of telomerase activity, which prevents the loss of DNA from chromosome ends and their integrity, may improve clinical outcome of HNSCC patients (McCaul et al., 2002). It is important to understand how DSBs repair efficiency may affect the sensitivity of tumor cells or patients whole body to chemo- and radiotherapy; thus, this study should be continued in order to draw further conclusion.
Conclusion
We found that the DNA repair capacity may vary depending on the damaging agents. These results suggest that head and neck cancer cells are highly sensitive to DNA damage; thus, treatment with DNA-reactive drugs and radiation might be considered as an effective therapy strategy. However, the data are still confusing, especially those concerning the advanced stage of this disease. Further research is needed to elucidate the molecular basis for successful therapy of HNSCC patients.
Footnotes
Acknowledgments
This work was supported by grant N301 099 32/3581 from Polish Ministry of Science and Higher Education.
Disclosure Statement
No competing financial interests exist.