
Abstract
Glucose-stimulated insulin gene transcription is mainly regulated by a 340-bp promoter region upstream of the transcription start site by beta-cell-enriched transcription factors Pdx-1, MafA, and NeuroD1. Previous studies have shown that histone H4 hyperacetylation is important for acute up-regulation of insulin gene transcription. Until now, only the histone acetyltransferase (HAT) protein p300 has been shown to be involved in this histone H4 acetylation event. In this report we investigated the role of the additional HAT proteins CREB binding protein (CBP), p300/CBP-associated factor (PCAF), and general control of amino-acid synthesis 5 (GCN5) in regulation of glucose-stimulated insulin gene transcription. Utilizing quantitative chromatin immunoprecipitation analysis, we demonstrate that glucose regulates the binding of p300, CBP, PCAF, and GCN5 to the proximal insulin promoter. siRNA-mediated knockdown of each of these HAT proteins revealed that depletion of p300 and CBP leads to a drastic decrease in histone H4 acetylation at the insulin promoter and in insulin gene expression, whereas knockdown of PCAF and GCN5 leads to a more moderate decrease in histone H4 acetylation and insulin gene expression. These data suggest that high glucose mediates the recruitment of p300, CBP, PCAF, and GCN5 to the insulin promoter and that all four HATs are important for insulin gene expression.
Introduction
Histone modifications in the underlying chromatin of the insulin promoter are important for both epigenetic maintenance of euchromatin structure and for the rapid regulation of glucose-stimulated insulin gene transcription (Chakrabarti et al., 2003; Mosley and Ozcan, 2003; Deering et al., 2009). Stable epigenetic marks for the insulin promoter are histone H3-Lys4 methylation and H3 Lys9,14 acetylation, which are enriched in the fully differentiated beta-cell (Chakrabarti et al., 2003). The islet-enriched methyltransferase Set7/9 maintains beta-cell identity by maintaining euchromatin structure of beta-cell genes, including the insulin promoter and coding region, by promoting histone H3-Lys4 mono- and di-methylation (Francis et al., 2005; Deering et al., 2009). Similarly, histone H3 Lys9,14 acetylation of the promoter is also enriched in a glucose-independent manner, indicating that both histone H3 acetylation and methylation function as epigenetic marks of the active insulin gene in the beta-cell.
Importantly, glucose stimulation of insulin gene transcription correlates with hyperacetylation of histone H4-Lys5,8,12,16 at the insulin promoter. Under high glucose conditions histone H4 acetylation is catalyzed by the histone acetyltransferase (HAT) p300, which is recruited to the proximal insulin promoter through a direct protein–protein interaction with Pdx-1 (Peshavaria et al., 2000; Mosley et al., 2004; Mosley and Ozcan, 2004; Wang et al., 2007). In the reciprocal regulation under low glucose conditions, a fraction of Pdx-1 remains bound to the insulin promoter and recruits the histone deacetylases HDAC-1 and HDAC-2, resulting in histone H4 hypoacetylation and downregulation of insulin gene transcription (Mosley and Ozcan, 2004). From these observations, Pdx-1 appears to act as a glucose-regulated molecular switch at the insulin promoter to regulate chromatin structure under appropriate glucose conditions.
Currently, only p300 has been implicated in regulation of histone acetylation at the insulin proximal promoter. However, several HAT proteins have been shown to interact with beta-cell-enriched transcription factors, including Pdx-1, MafA, and NeuroD1/Beta2. In addition to interacting with p300, Pdx-1 has also been shown to directly interact with the HAT CREB binding protein (CBP) through the N-terminal activation domain of Pdx-1 (Asahara et al., 1999). Furthermore, the HAT p300/CBP-associated factor (PCAF) has also been shown to play a role in regulating the function of beta-cell-enriched transcription factors. The acetylation of NeuroD1/Beta2 by PCAF enhances its DNA-binding capacity (Qiu et al., 2004). Furthermore, PCAF was shown in a recent study to enhance MafA protein stability through a direct protein–protein interaction (Rocques et al., 2007). However, the exact role of these HAT proteins in insulin gene transcription and pancreatic beta-cell function remains to be investigated.
While p300, CBP, and PCAF have been implicated to directly or indirectly to be important for beta-cell gene transcription, there is no information on the function of general control of amino-acid synthesis 5 (GCN5) in beta cells. GCN5, a paralog of PCAF, has been shown to play a central role controlling glucose metabolism in liver (Liu and Montminy, 2006). GCN5 responds to the nutrient state of the liver cell and directly regulates the transcription of gluconeogenic genes by acetylating and inhibiting Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC) (Lerin et al., 2006; Kelly et al., 2009; Caton et al., 2010; Dominy et al., 2010).
In this study we demonstrate that glucose stimulates insulin gene transcription by inducing the recruitment of p300, CBP, PCAF, and GCN5 to the insulin promoter. Knockdown of these HATs using specific siRNAs causes a reduction in hyperacetylation of histone H4 at the insulin promoter leading to decreased insulin gene transcription. These data suggest that the recruitment of HATs and hyperacetylation of histone H4 at the insulin promoter are important events in initiation of insulin gene transcription.
Materials and Methods
Cell culture
MIN6 cell passages between 21 and 25 were cultured as previously described (Miyazaki et al., 1990). Glucose regulation of HAT binding was studied by incubating MIN6 cells in 2.5 mM glucose overnight followed by stimulation with 2.5 mM or 25 mM glucose for 2 h before performing the described experiments.
Preparation of protein extracts and western blotting
Total lysate was extracted from MIN6 cells with standard lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.1%NP-40, 5 mM ethylenediaminetetraacetic acid, and protease/phosphatase inhibitors). Western blot analysis of whole cell lysate was conducted as previously described (Vanderford et al., 2007). Antibodies used are p300 (Santa Cruz, sc-584), CBP (Santa Cruz, sc-369), GCN5 (sc-6303), PCAF (Santa Cruz, sc-12124), and tetra-acetyl histone H4 (Upstate, 06-598). Densitometric analysis of western blots was conducted using Kodak 1D Image Analysis software. Relative quantification of band intensities was normalized to beta-actin loading controls for each sample and calculated as fold difference relative to scrambled siRNA control, which was set to a value of 100%.
Chromatin immunoprecipitation
For each sample, 2–5×106 MIN6 cells were processed for chromatin immunoprecipitation (ChIP) analysis as previously described with modifications (Chakrabarti et al., 2003; Mosley and Ozcan, 2003, 2004). In brief, cells were cross-linked in serum-free DMEM containing 1% formaldehyde for 10 min at room temperature and quenched with 10 mM glycine. Cell lysates were sonicated for 6 pulses (10 s each) using a Sonics Vibra-Cell Ultrasonic Processor at 50% amplitude setting. Lysate was pre-cleared with 30uL salmon-sperm blocked protein A or protein G sepharose beads for 1 h at 4°C with rotation. Lysate sample (5%) was set aside as total input. Samples were divided into two sample sets and used for immunoprecipitation with specific antibodies or with mouse, rabbit, or goat immunoglobulin G (IgG; 5 μg) as a negative control for nonspecific binding for 8–12 h at 4°C with rotation. Thirty microliters of salmon sperm-blocked protein A or protein G beads was added to the diluted lysate and rotated at 4°C for 1 h. Immune complexes were collected by centrifugation and washed. Protein-DNA cross-links were reversed by incubating with 200 mM NaCl at 65°C overnight. Samples were treated with RNAse (1 μL of 10 mg/mL per sample), followed by Proteinase K (10 μL of 10 mg/mL per sample). DNA was purified using the QIAQuick PCR Purification Kit (Qiagen) according to manufacturer's protocols.
Real-time polymerase chain reaction quantitation of ChIP-purified promoter fragments
Quantitative analysis of ChIP-purified DNA was conducted using real-time polymerase chain reaction (PCR) on an Mx4000 instrument (Stratagene) (Christenson et al., 2001; Chakrabarti et al., 2002; Mosley and Ozcan, 2004b). Real-time PCR amplification was performed using SYBR Green QPCR Master Mix (Stratagene) according to manufacturer's protocols. PCR fragments were observed on a polyacrylamide gel electrophoresis to confirm the presence of a single PCR band migrating at the correct size.
Ct values for each sample were normalized to total input DNA and IgG controls using the formula: 2[(Ct IgG−Ct Input)−(Ct Ab−Ct Input)], where Ab=p300, CBP, PCAF, GCN5, or tetra-acetyl histone H4. This calculation determines the fold enrichment of DNA in experimental samples over their corresponding IgG control, which accounts for background/nonspecific signal. For siRNA experiments, scrambled siRNA control values were set to 100%, and the corresponding experimental samples were expressed as a percentage of scrambled. This allowed for comparison among sample sets while normalizing for variations of ChIP immunoprecipitation efficiency among independent experiments. The primers for the mouse Ins2 promoter are 5′ AGGGCCCCTTGTTAAGACTCTAA 3′ and 5′ ACTGGGTCCCCACTACCTTTAT 3′.
Real-time (RT) PCR analysis
Quantitative PCR (qPCR) reactions were conducted on the Mx4000 real-time PCR instrument (Stratagene) as previously published (Mosley and Ozcan, 2004; Finlin et al., 2005; Vanderford et al., 2007). Cells were treated according to experimental conditions before harvesting of mRNA. RNA was isolated using the RNeasy Mini Kit (Qiagen) according to manufacturer's protocols. Isolated RNA was then treated with DNase I (Sigma) and used for cDNA synthesis with the Enhanced Avian HS RT-PCR Kit according to manufacturer's protocols. Insulin II pre-mRNA was amplified using SYBR Green qPCR master mix (Stratagene) with the following primers: 5′ GGGGAGCGTGGCTTCTTCTA 3′ and 5′ GGGGACAGAAT TCAGTGGCA 3′ as published previously (Evans-Molina et al., 2007).
siRNA-mediated gene silencing
One microgram of each siRNA oligo for every 1×106 cells was electroporated into MIN6 cells using the Nucleofector Device (Lonza Corp., catalog #VCA-003) according to manufacturer's protocols. Cells were then incubated in DMEM containing 25 mM glucose and 15% fetal bovine serum (FBS). After 12 h incubation, cells were switched to DMEM containing 25 mM glucose, 15% FBS, and antibiotic/antimycotic solution for an additional 36–40 h. Cells were then processed for mRNA harvesting, western blot, or ChIP analysis. The predesigned siRNA oligos (Ambion) used are CBP (5′ CCU AUCCGAGCAAACAUTT 3′), p300 (5′ GGCUUGACUUC UCCAAACATT 3′), PCAF (5′ GGAGUCCUGUAAAUGCA AUTT 3′), GCN5 (5′ GGAUCCUGAUCAGCUAUAUTT 3′), and scramble control siRNA #1 (Ambion). All siRNA oligos were confirmed to be sequence specific for the intended target protein.
Statistical analysis
All quantifications represent the mean of at least three independent experiments±standard deviation. Statistical significance was determined using a two-tailed Student's t-test. Sample sets with a p-value<0.05 were deemed statistically significant.
Results
Glucose stimulates histone H4 acetylation at the insulin promoter
It has been previously shown that glucose regulates the initiation of insulin gene transcription. Consistent with this, we found that 1 h exposure of MIN6 cells to 25 mM glucose increased Insulin II pre-mRNA levels by more than 15-fold compared to 2.5 mM glucose incubated cells (Supplementary Fig. S1; Supplementary Data are available online at
p300 and CBP are recruited to the insulin promoter and mediate histone H4 acetylation
Several published reports indicate that p300 is an essential co-regulator of insulin gene transcription and is recruited to the insulin gene promoter via the transcription factor Pdx-1 (Mosley et al., 2004). To further investigate the specific role of p300 in regulation histone H4 acetylation and insulin gene transcription in MIN6 cells, ChIP analysis of p300 binding and histone H4 acetylation at the mouse proximal Ins2 promoter was conducted using specific primers. ChIP analysis of MIN6 cells treated with 2.5 mM or 25 mM glucose showed about a twofold enrichment of p300 at the insulin promoter under high glucose conditions (Fig. 1A, Supplementary Fig. S3A). To assess the role of p300 in insulin gene transcription under 25 mM glucose conditions, p300 protein levels were depleted by 80% in MIN6 cells using a specific siRNA (Fig. 1B). siRNA-treated MIN6 cells were preincubated with 2.5 mM glucose overnight followed by 25 mM glucose treatment for 2 hours before ChIP analysis of histone H4-hyperacetylation. Ablation of p300 in MIN6 cells by RNAi resulted in an almost complete loss of detectable levels of histone H4 hyperacetylation at the proximal promoter region under high glucose conditions (Fig. 1C, Supplementary Fig. S4A). Furthermore, quantification of insulin II pre-mRNA transcript levels by quantitative real time (qRT)-PCR in MIN6 cells depleted for p300 indicated a 70% decrease in insulin gene transcription (Fig. 1D).
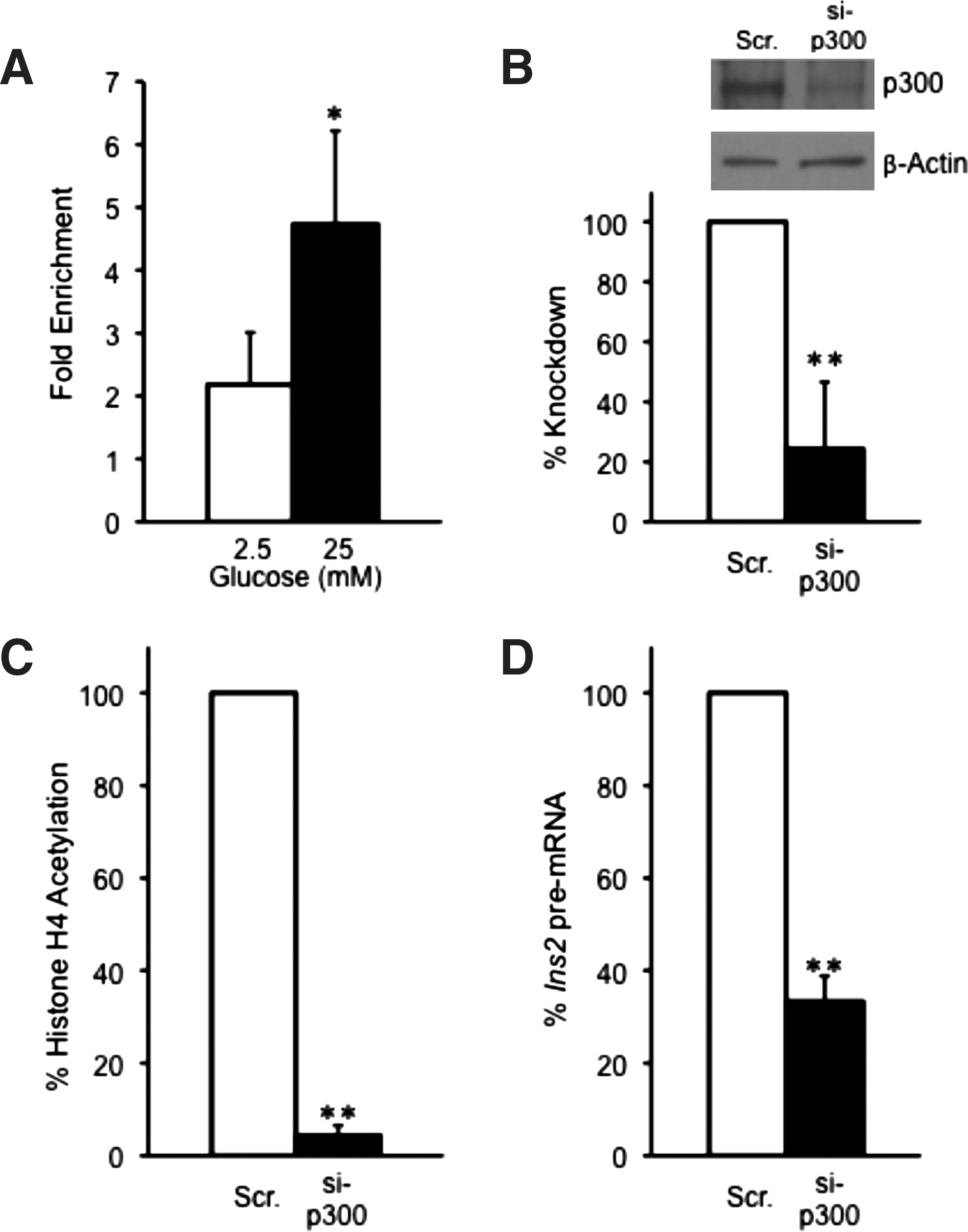
Glucose mediates p300 binding to the Ins2 promoter to regulate histone H4 hyperacetylation. MIN6 cells were preincubated ∼12 h with 2.5 mM glucose followed by 2-h stimulation with either 2.5 or 25 mM glucose. Cells were processed for ChIP analysis for p300 association with the Ins2 proximal promoter
In addition to p300, CBP has also been previously reported to interact with the N-terminal activation domain of Pdx-1 (Asahara et al., 1999). However, no prior information about the role of CBP in histone H4 acetylation at the insulin promoter is available. ChIP analysis of MIN6 cells treated with either 2.5 mM or 25 mM glucose indicated that CBP binds to the insulin promoter under both low and high glucose conditions. However, there was about a threefold enrichment of CBP at the insulin promoter under 25 mM glucose conditions, indicating that high glucose stimulates CBP binding (Fig. 2A; Supplementary Fig. S3B). siRNA-mediated ablation of CBP levels by 80% resulted in an 80% loss of glucose-stimulated histone H4 hyperacetylation of the proximal promoter region (Fig. 2B, C, Supplementary Fig. S4B). Furthermore, CBP depletion caused a ∼65% reduction in insulin II pre-mRNA levels after stimulation with high glucose (Fig. 2D). These data, taken together, suggest that p300 and CBP both are required for histone H4 acetylation and insulin gene transcription initiation upon glucose stimulation.
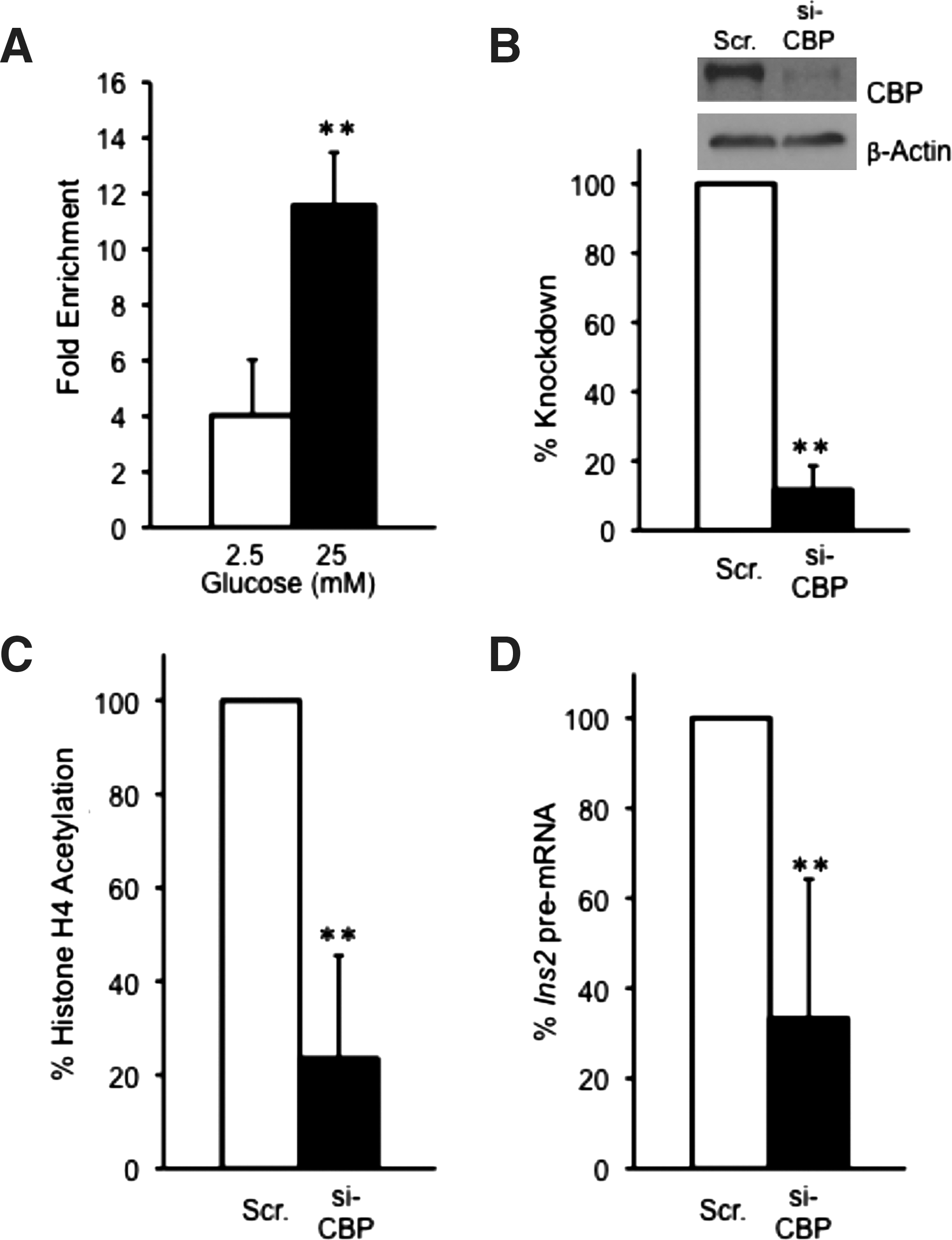
CBP is associated with the Ins2 promoter and regulates insulin gene transcription.
PCAF associates with the insulin II promoter and mediates histone H4 acetylation
Previous data indicate that PCAF can enhance the DNA-binding capacity of BETA2/NeuroD1 and promote MafA stability by inhibiting the ubiquitination of MafA (Qiu et al., 2004; Rocques et al., 2007). However, it is unclear if PCAF is involved in insulin gene transcription. To investigate whether PCAF is recruited to the insulin promoter, we conducted ChIP analysis in MIN6 cells under 2.5 and 25 mM glucose conditions. As shown in Figure 3A, PCAF is recruited to the insulin promoter and there is an approximately sixfold enrichment of PCAF at the insulin promoter on 25 mM glucose compared to 2.5 mM glucose, where PCAF association was barely detectable (Supplementary Fig. S3C). To specifically analyze the effects of PCAF on histone H4 hyperacetylation and insulin gene transcription, PCAF was ablated by about 70% in MIN6 cells via siRNA knockdown before ChIP analysis (Fig. 3B). Loss of PCAF protein resulted in a 60% reduction in histone H4 hyperacetylation. However, depletion of PCAF by siRNA led only to a 20% reduction in insulin II pre-mRNA levels (Fig. 3C, D, Supplementary Fig. S4C). These data suggest that although PCAF is associated with the insulin promoter at high glucose levels and contributes to some extent to histone H4 hyperacetylation, its effect on initiation of insulin gene transcription is minimal compared to p300 or CBP.
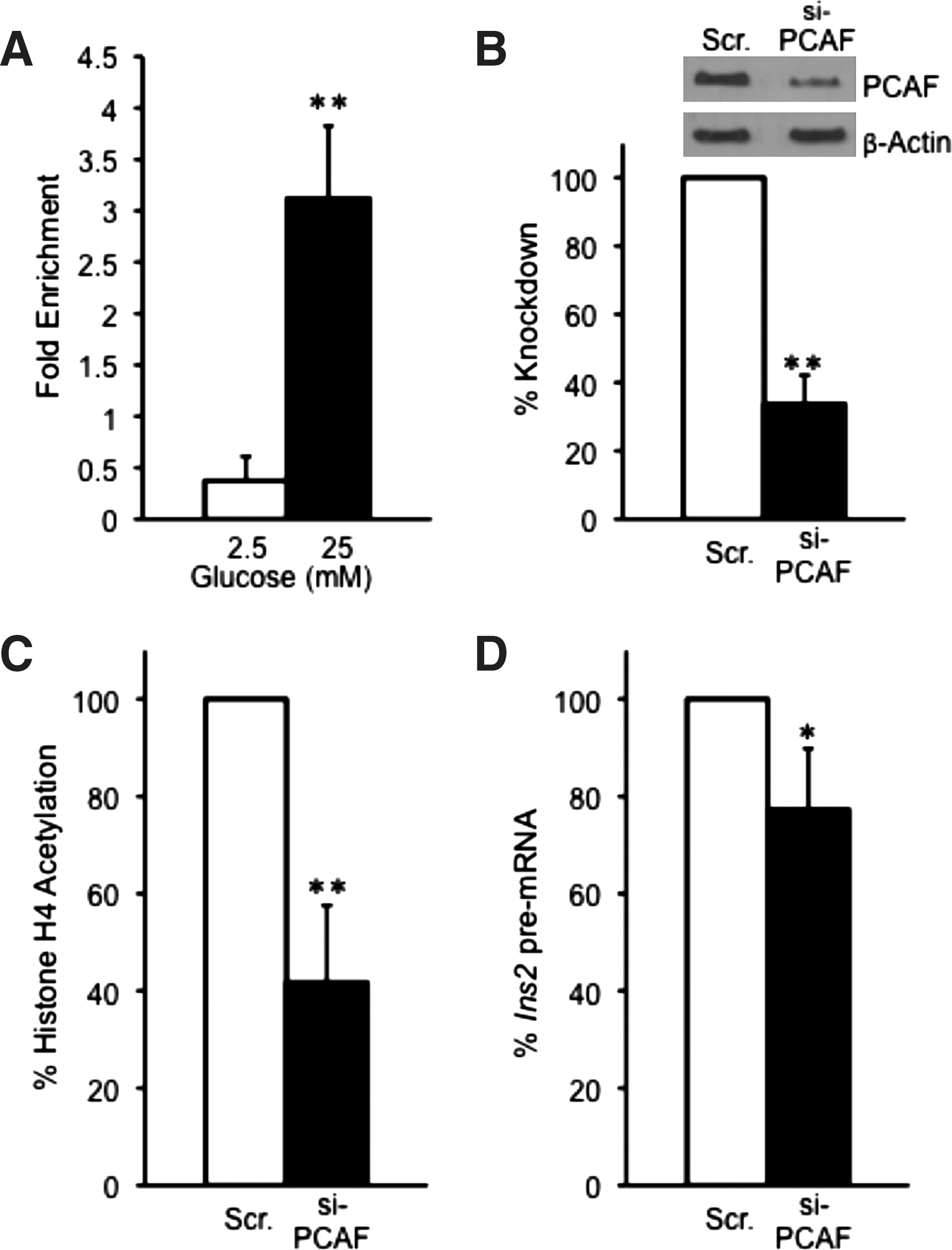
Glucose causes the recruitment of PCAF to the Ins2 promoter to regulate histone H4 hyperacetylation.
GCN5 is recruited to the insulin promoter and is required for glucose induction of insulin gene transcription
Several recent reports link GCN5 to regulation of glucose metabolism in liver by responding to nutrient conditions and regulating the transactivation potential of the transcription factor PGC1alpha at gluconeogenic gene targets (Lerin et al., 2006; Kelly et al., 2009; Caton et al., 2010; Dominy et al., 2010). Thus, we tested whether GCN5 regulates insulin gene transcription in response to glucose. As shown in Figure 4A, ChIP analysis with the GCN5 antibody indicates that GCN5 binds to the insulin promoter in a glucose-regulated manner. Binding of GCN5 to the insulin II promoter is about sevenfold increased on high versus low glucose conditions, where binding was almost undetectable (Fig. 4A, Supplementary Fig. S3D). siRNA-mediated silencing of GCN5 caused a 60% reduction in GCN5 protein levels (Fig. 4B). Surprisingly, however, loss of GCN5 did not cause a statistically significant reduction of histone H4 hyperacetylation, but resulted in a 40% decrease in insulin II pre-mRNA levels under high glucose conditions (Fig. 4C, D, Supplementary Fig. S4D). These data indicate that GCN5 is to some extent important for insulin gene transcription, but this is independent of histone H4 hyperacetylation.
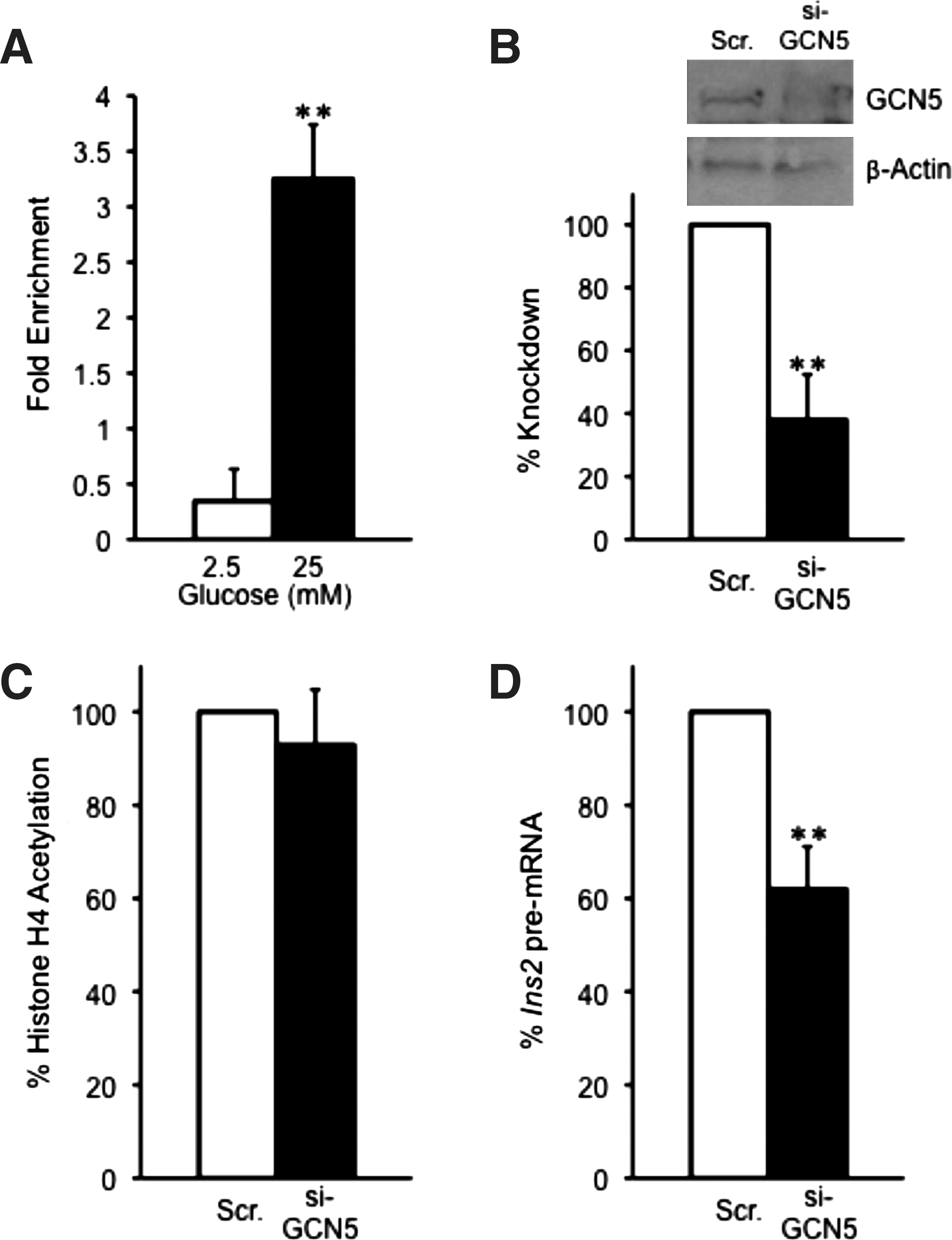
GCN5 is recruited to the Ins2 promoter in a glucose-dependent manner, but depletion of GCN5 has no effect on histone H4 acetylation.
Discussion
There is accumulating evidence linking histone modification events to regulation of glucose homeostasis. However, the exact mechanisms by which chromatin modifications regulate gene expression to control glucose homeostasis are poorly understood. In the pancreatic beta-cell, reversible hyperacetylation of histone H4-Lys5,8,12,16 has been shown to contribute to acute glucose-stimulated insulin gene transcription (Mosley and Ozcan, 2003; Evans-Molina et al., 2007). Previous data suggest that under high glucose conditions, histone H4 hyperacetylation occurs in a process dependent on an interaction between p300 and the N-terminal activation domain of Pdx-1 (Mosley et al., 2004a). Conversely, under low glucose conditions, it has been shown that Pdx-1 interacts with HDAC-1 and HDAC-2 to downregulate insulin transcription by histone H4 deacetylation (Mosley and Ozcan, 2004b). A recent report demonstrates that the beta-cell-specific transcription factor insulin-associated antigen-1 (INSM1/A-1) is also able to mediate histone H4 acetylation at the insulin promoter and thereby promotes ductal cell transdifferentiation (Zhang et al., 2010). Until now, only p300 has been implicated as a HAT protein required for glucose-regulated histone H4 hyperacetylation at the insulin promoter (Mosley et al., 2004). This study provides new insight into the HATs that participate in this process. Using quantitative ChIP analysis, we found that all four HATs (p300, CBP, PCAF, and GCN5) are recruited to the insulin promoter in a glucose-dependent manner in the MIN6 cell line. Depletion of all four HATs causes a reduction in insulin gene transcription to varying extent.
While depletion of p300, CBP, and PCAF leads to a significant reduction in histone H4 hyperacetylation, knockdown of PCAF reduced insulin gene transcription only by 20%. Thus, in this case there was no exact correlation between histone H4 acetylation and insulin II pre-mRNA levels. It is likely that a 40% reduction in histone H4 acetylation level as observed after depletion of PCAF is still sufficient to mediate the assembly of the transcription initiation complex at the insulin promoter. However, it is still surprising that the depletion of PCAF has only a minimal effect on insulin gene transcription given that PCAF has been shown to regulate the function of the insulin gene transcription factors NeuroD1/Beta2 and MafA. PCAF has been demonstrated to acetylate NeuroD1/Beta2 and to enhance its DNA-binding capacity (Qiu et al., 2004). Furthermore, previous data indicate that MafA ubiquitination and degradation is prevented by its interaction with PCAF (Rocques et al., 2007), though no data currently exist on MafA acetylation. The finding that depletion of PCAF only slightly affects insulin gene transcription may be explained by redundancy of HAT function. In the absence of PCAF, may be one or more of the remaining HATs replace PCAF function.
Another surprising result was that knockdown of GCN5 caused a 40% reduction in insulin gene expression without affecting histone H4 acetylation levels at the insulin promoter. This result indicates that GCN5 does not directly participate in acetylation of histone H4 and may regulate insulin gene transcription by acetylating of transcription factors that directly bind the proximal insulin promoter, such as Pdx-1, MafA, and/or NeuroD1/Beta2. Indeed, in liver GCN5 has been demonstrated to regulate gluconeogenic gene expression by acetylation of the transcriptional co-activator PGC1 alpha in response to the energy status of the cell (Lerin et al., 2006; Liu and Montminy, 2006; Kelly et al., 2009; Dominy et al., 2010). Alternatively, histone H4 acetylation may only serve as a marker for recruitment of other transcriptional regulators to the insulin promoter and may not be directly involved in insulin gene transcription.
Our study suggests that glucose as a nutrient regulates the recruitment of HATs to the insulin gene promoter, thereby linking histone acetylation to nutrient status of the cell. A likely mechanism by which histone acetylation levels could be altered in dependence of the nutrient or energy state of the cell is via the intracellular pools of acetyl-CoA (Jeninga et al., 2010). Interestingly, a recent report indicates that in mammalian cells, metabolism is directly linked to histone acetylation by the activity of a nuclear fraction of the enzyme ATP citrate lyase (ACL). This nuclear fraction of ACL uses glucose-derived citrate to produce a nuclear pool of acetyl-CoA, which then serves as a localized substrate for HAT activity (Wellen et al., 2009). Based on this observation, it is likely that a similar mechanism is employed in beta cells to rapidly respond to glucose metabolism for the upregulation of histone H4 acetylation at the insulin gene promoter and other glucose-regulated targets. Additional studies are required to determine if nuclear ACL activity is required for glucose-stimulated histone H4 acetylation at the insulin promoter and for insulin gene transcription.
Footnotes
Acknowledgments
This work was supported by grants R01DK067581 from NIH/NIDDK, P20RR020171 from NIH/NCRR, and 1-05-CD-15 from the American Diabetes Association and by a Predoctoral Fellowship from the American Heart Association (0715502B) to M.L.S.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.