
Abstract
Polymorphisms such as single-nucleotide polymorphisms (SNPs) and insertions/deletions (Indels) can be associated with phenotypic traits and be used as markers for disease diagnosis. Identification of these genetic variations within laboratory mice is crucial to improve our understanding of the genetic background of the mice used for research. As part of a positional cloning project, we sequenced six genes (Mettl16, Evi2a, Psmd11, Cct6d, Rffl, and Ap2b1) within a 6.8-Mb domain of mmu chr 11 in the C57BL/6J and 129S6/SvEvTac inbred strains. Although 129S6/SvEvTac is widely used in the mouse community, there is very little current (or projected future) sequence information available for this strain. We identified 6 Indels and 21 novel SNPs and confirmed genotype information for 114 additional SNPs in these 6 genes. Mettl16 and Ap2b1 contained the largest numbers of variants between the C57BL/6J and 129S6/SvEvTac strains. In addition, we found five new SNPs between 129S6/SvEvTac and 129S1/SvImJ within the Ap2b1 locus. Although we did not detect differences between C57BL/6J and 129S6/SvEvTac within Evi2a, this locus contains a relatively high SNP density compared with the surrounding sequence. Our study highlights the genetic differences among three inbred mouse strains (C57BL/6J, 129S6/SvEvTac, and 129S1/SvImJ) and provides valuable sequence information that can be used to track alleles in genomics-based studies.
Introduction
Different types of polymorphisms are generally created by different kinds of alteration events. For example, SNPs, the most common types of polymorphisms, result from a single base substitution. The most common SNP in the mammalian genome is a C to T transition, whereby a methylated cytosine is spontaneously deaminated to form a thymine base (Miller et al., 2001). As this change is not recognized by DNA repair enzymes, the SNP segregates within the population unless it is under selection, is lost, or by chance randomly fixes. SNP variation in protein-coding genes and in other functionally constrained regions of the genome, such as promoters, enhancers, miRNAs, and other noncoding sequences, contributes significantly to phenotypic variation (Morin, 2004; Sethupathy et al., 2007; Bartel, 2009).
Indels are often, but not always, present in highly repetitive sequences (Chen et al., 2009). They are abundantly distributed across the genome, but not as common as SNPs (Vali et al., 2008). Both of these types of genetic variations can be used as tools in genotyping as well as discovery of disease- or trait-related genes by directly sequencing SNPs or identifying Indels based on size separation or sequence.
Many mouse polymorphism databases are available, such as the Mouse Genome Informatics (MGI) strains, SNPs and Polymorphisms database (
While conducting a positional cloning project to identify induced variants from an N-ethyl-N-nitrosourea (ENU) mutagenesis screen, we detected several 129S6/SvEvTac polymorphisms in potential candidate genes. This ENU mutagenesis screen was performed on C57BL/6J males, which were bred to 129S6/SvEvTac animals carrying an inversion chromosome for the targeted region (Kile et al., 2003; Hentges et al., 2006). The mouse reference sequence is based on C57BL/6J, which is one of the most widely used inbred mouse strains for the generation and analysis of transgenic mice and disease models (Al-Hasani et al., 2004; Tang et al., 2008). 129S6/SvEvTac and 129S1/SvImJ have been used frequently for gene knockout studies (Hibma et al., 2007; Kuo et al., 2010). 129S1/ImJ is one of the 17 additional mouse strains being sequenced by the Sanger Center (Turner et al., 2009). However, there is very little polymorphism data for 129S6/SvEvTac. As a result, it is important to develop SNP and other polymorphism detection assays to help identify 129S6/SvEvTac alleles to facilitate genotyping. Here we provide SNP and Indel information for six genes located on chromosome 11 between these three commonly used laboratory mouse strains.
Materials and Methods
We isolated genomic DNA from mouse tail biopsies from 129S6/SvEvTac and C57BL/6J animals; 129S1/SvImJ DNA was purchased from the Jackson Laboratory. Exons from each gene were sequenced individually. Each 600–800-bp polymerase chain reaction (PCR) amplicon contained one or more exons plus upstream and downstream flanking sequence. Exons larger than 800 bp were sequenced by multiple overlapping sequencing reactions. All amplicons were sequenced in both directions using Big Dye v3.1 chemistry (Applied Biosystems, Foster City, CA). Primers are listed in Table 1.
We analyzed the data using Sequencher 4.9 (Gene Codes, Ann Arbor, MI) to identify polymorphisms among the various strains used in this study. The sequences from different samples were assembled automatically and then entered into a BLAT search against the UCSC genome database (
Results
We analyzed Mettl16, Evi2a, Psmd11, Cct6d, Rffl, and Ap2b1 on mouse chromosome 11. Chromatographs for representative SNPs within each locus are shown (Fig. 1). We only detected differences between C57BL/6J and 129S6/SvEvTac in Mettl16 and Ap2b1. However, we summarized SNP information for sequenced genes.
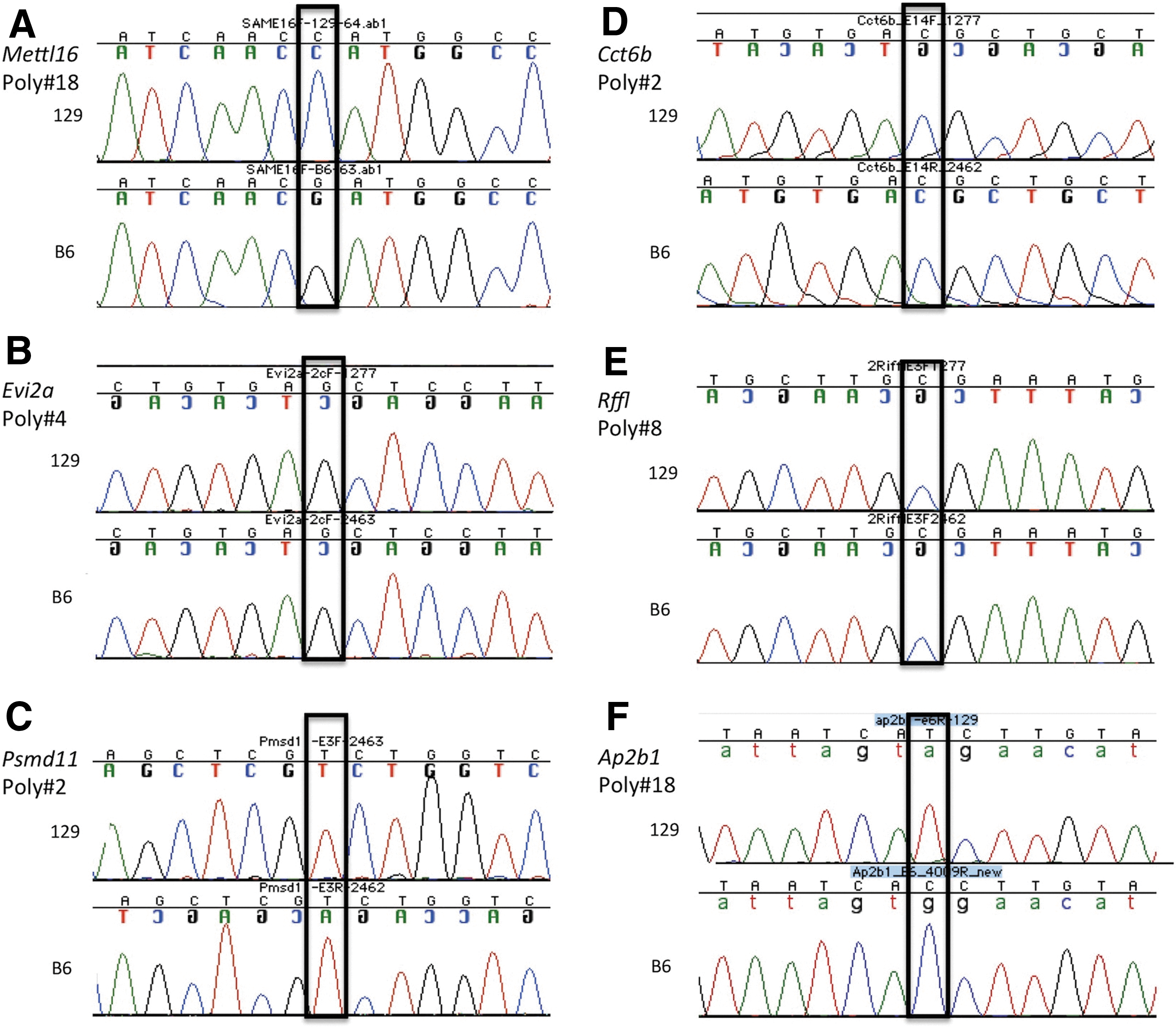
Representative chromatographs for Mettl16
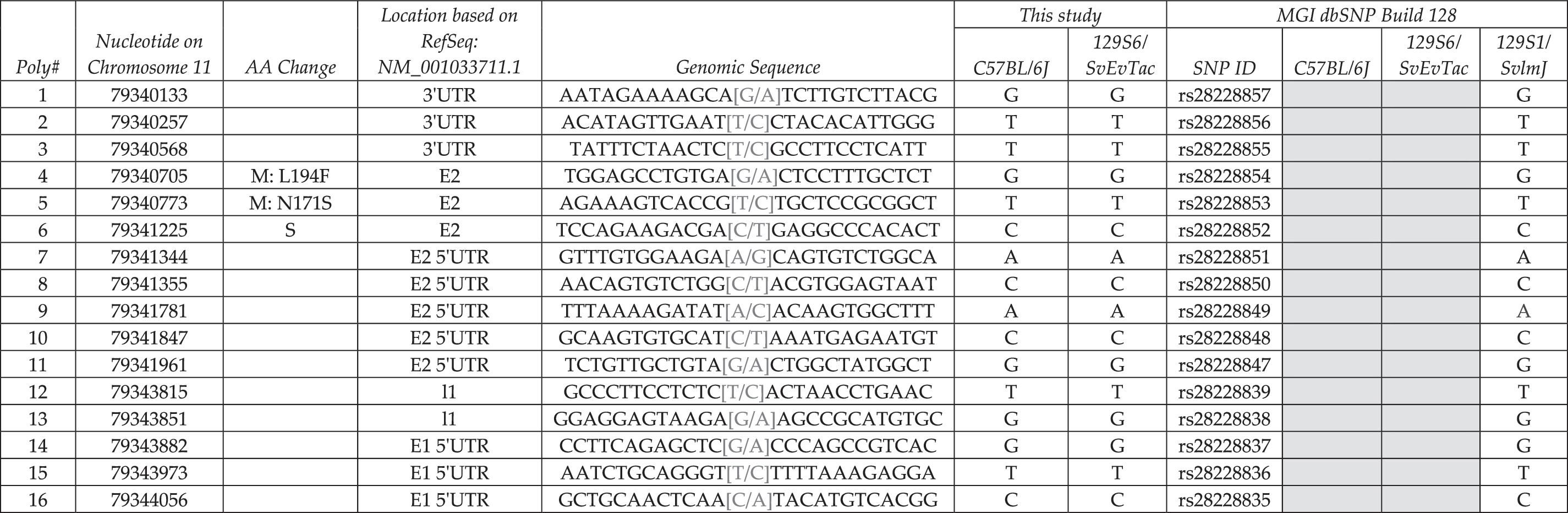
The RefSeq annotated transcript for Methyltransferase like 16 (Mettl16), the most proximal gene in the group, is expressed from the Watson strand and contains 10 exons; 4 additional exons are predicted based on Ensembl annotations. We sequenced all 14 exons and identified 18 variants (Table 2). Although C57BL/6J is the mouse reference sequence, the dbSNP database lacks significant C57BL/6J information. Gray lettering denotes the variant, gray boxes indicate new dbSNP information, and bold lettering indicates novel sequence variants between C57BL/6J and 129S6/SvEvTac; all new variants have been deposited into the dbSNP database.

Column 1 denotes the relative number based on chromosome position and column 2 indicates the actual nucleotide that is changed based on NCBI Build 37 at the UCSC and Ensembl genome browsers. Location of the variant within the locus (i.e., upstream, 5′ and 3′ UTR, exonic and intronic alleles) is indicated in column 3, for example, intron 8 is denoted I8. We provide upstream and downstream sequence information in column 4, and columns 5 and 6 summarize sequencing data from this study. dbSNP ID number is presented in column 7, and dbSNP genotyping data are shown in columns 8–10; variants without dbSNP IDs are novel alleles. Gray shade indicates no data available in current database. Dark shading indicates distinction between C57BL/6J and 129S6/SvEvTac. Boldface indicates new SNP or indel. Gray letters indicate new information for 129S1/SvlmJ. Gray-colored sequence denotes the variant.
SNP, single nucleotide polymorphism; indel, insertions/deletions polymorphisms; E, Exon; I, Intron; US, upstream; DS, downstream; 3′UTR, 3′ untranslated region; 5′UTR, 5′ untranslated region S, silent variant; M, missense change.
We identified two new SNPs (10 and 18) and Indels (11 and 14) within Mettl16. SNP 10 is a T to G transversion in intron 5, whereas SNP 18 is a G to C transversion located in intron 8. Indel 11 is a 3 bp insertion located within a string of Ts, and it is therefore impossible to determine the exact insertion site. Indel 14 is a 12 bp deletion in 129S6/SvEvTac. We confirmed the C57BL/6J genotype for polymorphisms 13, 16, and 17 and updated the dbSNP database for 13 additional C57BL/6J and all 16 129S6/SvEvTac alleles. Within the Mettl16 locus, 129S6/SvEvTac and 129S6/SvImJ are concordant for all variants tested with both strains. Notably, six SNPs (10, 13, and 15–18) and two Indels (11 and 14) differ between 129S6/SvEvTac and C57BL/6J.
Ecotropic viral integration site 2A (Evi2a) is expressed from the Crick strand and contains two exons (Table 3). Table 3 is organized the same as Table 2, with the following exceptions: amino acid changes are found in column 3 and the remaining column numbers are shifted. We designed five amplicons for sequencing Evi2a, as exon 2 spans 2058 bp. We confirmed the 16 SNPs in the region and did not identify any additional alleles. However, we added genotyping information for C57BL/6J for all SNPs. Three SNPs (4–6) are located in exon 2; SNPs 4 and 5 cause missense changes (M), whereas SNP 6 is a silent variant (S).
Column 1 denotes the relative number based on chromosome position and column 2 indicates the actual nucleotide that is changed based on NCBI Build 37 at the UCSC and Ensembl genome browsers. Amino acid changes are listed in column 3. Location of the variant within the locus (i.e., upstream, 5′ and 3′ UTR, exonic and intronic alleles) is indicated in column 4, for example, intron 8 is denoted I8. We provide upstream and downstream sequence information in column 5, and columns 6 and 7 summarize sequencing data from this study. dbSNP ID number is presented in column 8, and dbSNP genotyping data are shown in columns 9–11; variants without dbSNP IDs are novel alleles. Gray shade indicates no data available in current database. Blue letters indicate new information for 129S1/SvlmJ.
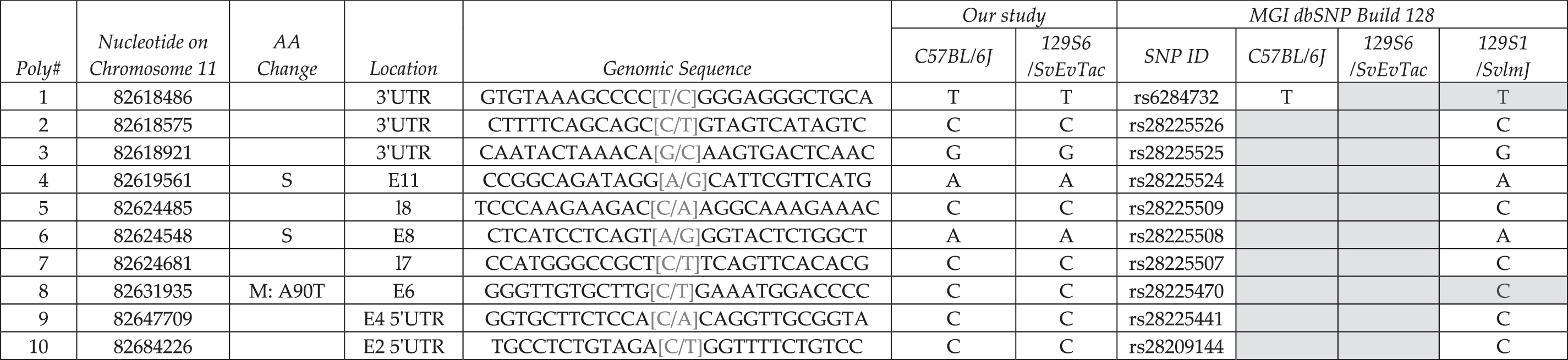
Proteasome 26S non-ATPase subunit 11 (Psmd11) codes from the Watson strand and contains 14 exons (Table 4). Table 4 has the same organization as Table 3. We designed 13 amplicons for sequencing. Exons 11 and 12 were amplified from one PCR product. We confirmed the four SNPs in the region and did not identify any additional alleles. However, we added genotyping information for C57BL/6J for all SNPs. SNP 2 causes a silent variant in exon 3.

Column 1 denotes the relative number based on chromosome position and column 2 indicates the actual nucleotide that is changed based on NCBI Build 37 at the UCSC and Ensembl genome browsers. Amino acid changes are listed in column 3. Location of the variant within the locus (i.e., upstream, 5′ and 3′ UTR, exonic and intronic alleles) is indicated in column 4, for example, intron 8 is denoted I8. We provide upstream and downstream sequence information in column 5, and columns 6 and 7 summarize sequencing data from this study. dbSNP ID number is presented in column 8, and dbSNP genotyping data are shown in columns 9–11; variants without dbSNP IDs are novel alleles. Gray shade indicates no data available in current database.
Chaperonin containing Tcp1, subunit 6b (zeta) (Cct6b) is expressed from the Crick strand and contains 14 exons (Table 5). Table 5 has the same organization as Table 2. We sequenced each exon and confirmed nine SNPs within this locus. SNP 1 is located downstream to Cct6b, whereas SNPs 2 and 3 are within the 3′UTR and should be identified from mRNA. Although we did not identify any additional alleles, we added genotyping information for C57BL/6J for nine SNPs.

Column 1 denotes the relative number based on chromosome position and column 2 indicates the actual nucleotide that is changed based on NCBI Build 37 at the UCSC and Ensembl genome browsers. Location of the variant within the locus (i.e., upstream, 5′ and 3′ UTR, exonic and intronic alleles) is indicated in column 3, for example, intron 8 is denoted I8. We provide upstream and downstream sequence information in column 4, and columns 5 and 6 summarize sequencing data from this study. dbSNP ID number is presented in column 7, and dbSNP genotyping data are shown in columns 8–10; variants without dbSNP IDs are novel alleles. Gray shade indicates no data available in current database.
Ring finger and FYVE like domain containing (Rffl) is expressed from the Crick strand, contains 11 exons, and produces 5 alternatively spliced products (Table 6). Table 6 is organized exactly the same as Table 3. We designed 13 amplicons for sequence analysis of Rffl; 3 span exon 11. To reduce complexity, we incorporated all splice variants into a single transcript. RefSeq NM_026097.3 contains exons 5–7 and 9–11. RefSeq NM_001164570.1 contains exons 2, 4, and 6–11. RefSeq NM_001007465.3 contains exons 1 and 6–11. RefSeq NM_001164569.1 contains exons 1, 2, and 6–11. RefSeq NM_001164571.1 contains exons 1, 6, 7, and 9–11. We confirmed 10 SNPs for this locus and found no variants between C56BL/6J and 129S6/SvEvTac. SNPs 9 and 10 are within the 5′UTR of exons 4 and 2, respectively. SNP 8 is a missense variant in exon 6, whereas SNPs 6 and 4 are silent changes in exons 11 and 8, respectively. SNPs 1–3 are located in the 3′UTR of Rffl. SNP 8 causes an Ala to Thr missense change at amino acid positions 90 (NM_001164569.1), 69 (NM_001164570.1), and 55 (NM_026097.3, NM_001007465.3 and NM_001164571.1) depending upon the RefSeq.
Column 1 denotes the relative number based on chromosome position and column 2 indicates the actual nucleotide that is changed based on NCBI Build 37 at the UCSC and Ensembl genome browsers. Amino acid changes are listed in column 3. Location of the variant within the locus (i.e., upstream, 5′ and 3′ UTR, exonic and intronic alleles) is indicated in column 4, for example, intron 8 is denoted I8. We provide upstream and downstream sequence information in column 5, and columns 6 and 7 summarize sequencing data from this study. dbSNP ID number is presented in column 8, and dbSNP genotyping data are shown in columns 9–11; variants without dbSNP IDs are novel alleles. Gray shade indicates no data available in current database. Blue letters indicate new information for 129S1/SvlmJ.
Adaptor-related protein complex 2, beta 1 (Ap2b1) is expressed from the Watson strand and contains 22 exons (RefSeq NM_001035854.2) (Table 7). Table 7 is organized exactly the same as Table 3. We designed 30 amplicons: 6 are necessary to span exon 22, 5 are necessary to span exon 12, and exons 8 and 9 are amplified from a single PCR reaction. We identified 80 SNPs and 4 Indels; 19 of the SNPs (28–32, 36, 37, 41–48, 50, 65, 66, and 84) and all Indels (4, 12, 27, and 80) are novel. SNPs 1–3 are located upstream of the transcriptional start site of the two RefSeq genes (NM_001035854.2 and NM_027915.3), but within the 5′UTR of the predicted Ensembl transcripts (ENSMUST00000018875). Variant 4 (a 6 bp insertion) is located within the 5′UTR of exon 1. The remaining three Indels (12, 27, and 80) are located in intron 3, intron 10, and the 3′UTR, respectively. SNP 32 causes a silent change in exon 11. The remaining novel SNPs are located within introns and the 3′UTR. Previously identified SNPs (33 and 56) lead to missense variants (F490L and A602T) in exons 11 and 14, respectively. Additionally expressed polymorphisms include exons 4, 7, 12, 13, 14, 17, and 20. We identified 5 variants between 129S6/SvEvTac and 129S1/SvImJ (blue boxes, 16, 33, 53, 60, and 82) and 46 differences between 129S6/SvEvTac and C57BL/6J (yellow boxes). Notably, SNP 33 leads to a missense variant between 129S6/SvEvTac and 129S1/SvImJ that could account for differences between these two distinct 129 strains.
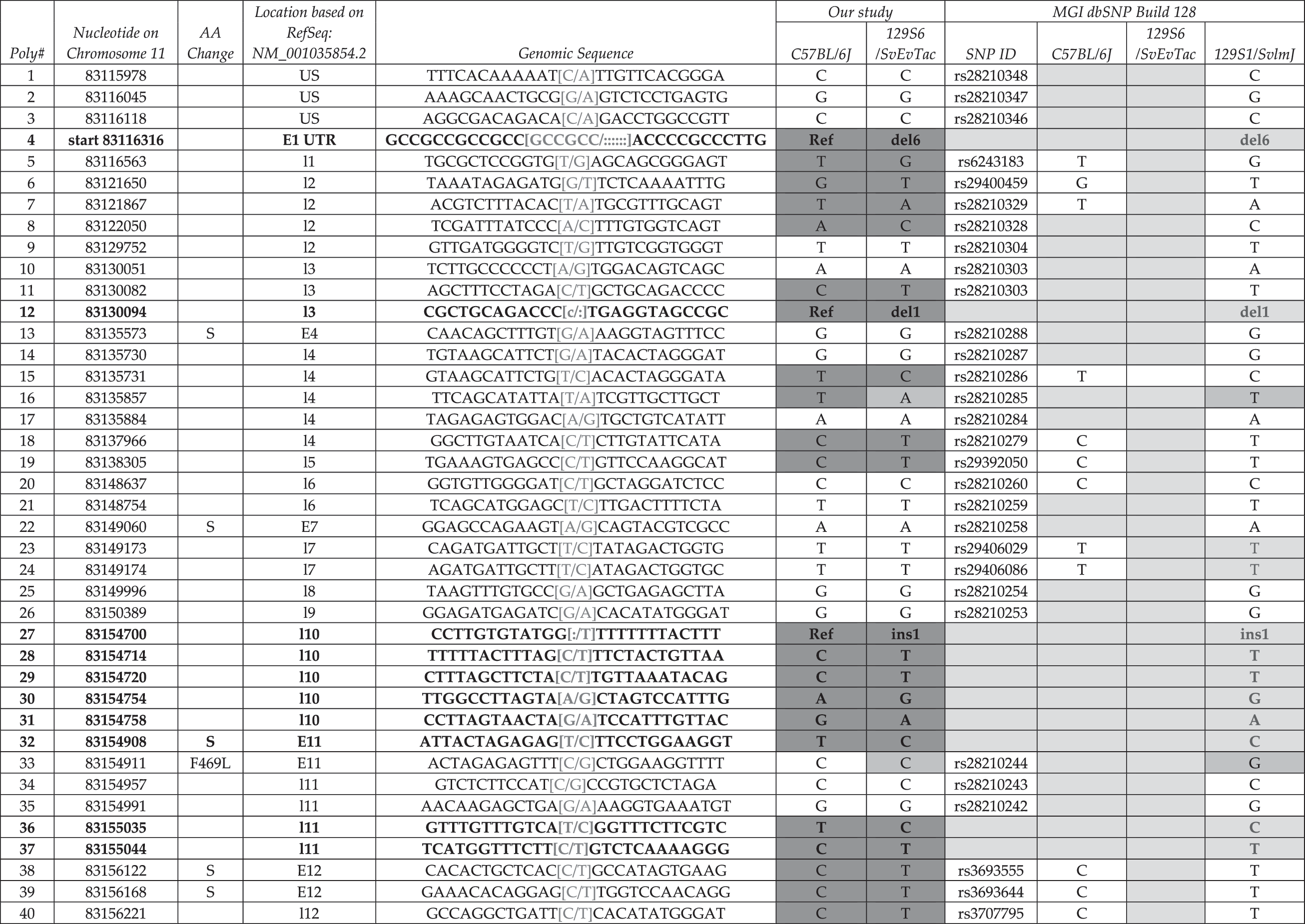
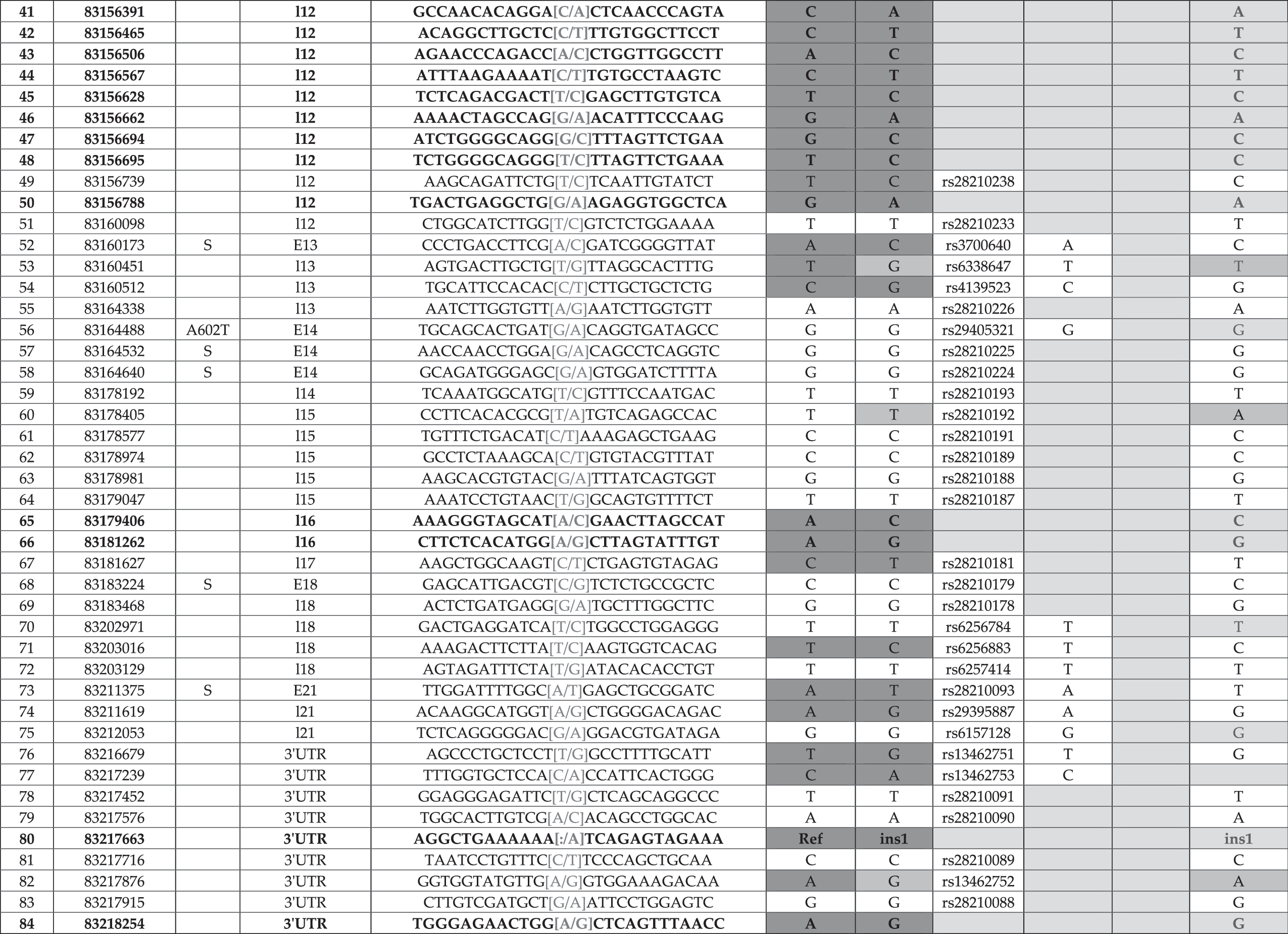
Column 1 denotes the relative number based on chromosome position and column 2 indicates the actual nucleotide that is changed based on NCBI Build 37 at the UCSC and Ensembl genome browsers. Location of the variant within the locus (i.e., upstream, 5′ and 3′ UTR, exonic and intronic alleles) is indicated in column 3, for example, intron 8 is denoted I8. We provide upstream and downstream sequence information in column 4, and columns 5 and 6 summarize sequencing data from this study. dbSNP ID number is presented in column 7, and dbSNP genotyping data are shown in columns 8–10; variants without dbSNP IDs are novel alleles. Gray shade indicates no data available in current database. Yellow shade indicates distinction between C57BL/6J and 129S6/SvEvTac. Boldface indicates new SNP or indel. Blue shade indicates distinction between 129S6/SvlmJ and 129S6/SvEvTac. Blue letters indicate new information for 129S1/SvlmJ.
The polymorphisms located in these six genes between 129S6/SvEvTac and C57BL/6J strain are summarized in Table 8. In total, we identified 6 Indels and 135 SNPs in all 6 genes; we are the first to report 21 of these SNPs and all Indels. Regarding our analyses, it becomes clear that the major distinctions between the 129S6/SvEvTac and C57BL/6J strains lie within Ap2b1 and Mettl16, whereas the variations between 129S6/SvEvTac and 129S1/SvImJ lie in Ap2b1. This suggests that the remaining four genes might be more highly conserved among the 129S6/SvEvTac, 129S1/SvImJ, and C57BL/6J strains.
UTR SNP indicates the number of SNPs within the untranslated regions.
The distinction indicates the number of differences in SNPs or indel between 129S6/SvEvTac and C57BL/6J.
The distinction indicates the number of differences in SNPs or indel between 129S6/SvEvTac and 129S1/SvImJ.
The distinction indicates the number of differences in SNPs or indel between C57BL/6J and 129S1/SvImJ.
We converted the current dbSNP information within this region into a heatmap that represents the polymorphism density across the region (Fig. 2). Dark areas contain more variants than light gray segments. It is clear that the six target genes all map to regions that contain high or intermediate numbers of SNPs. Interestingly, there are two blocks with a low SNP density. The first lies between Mettl16 and Evi2a and the second falls between Evi2a and Psmd11. Based on the SNP density, we predict that these two regions contain elements under selective pressure in the two mouse strains or harbor important functional domains.
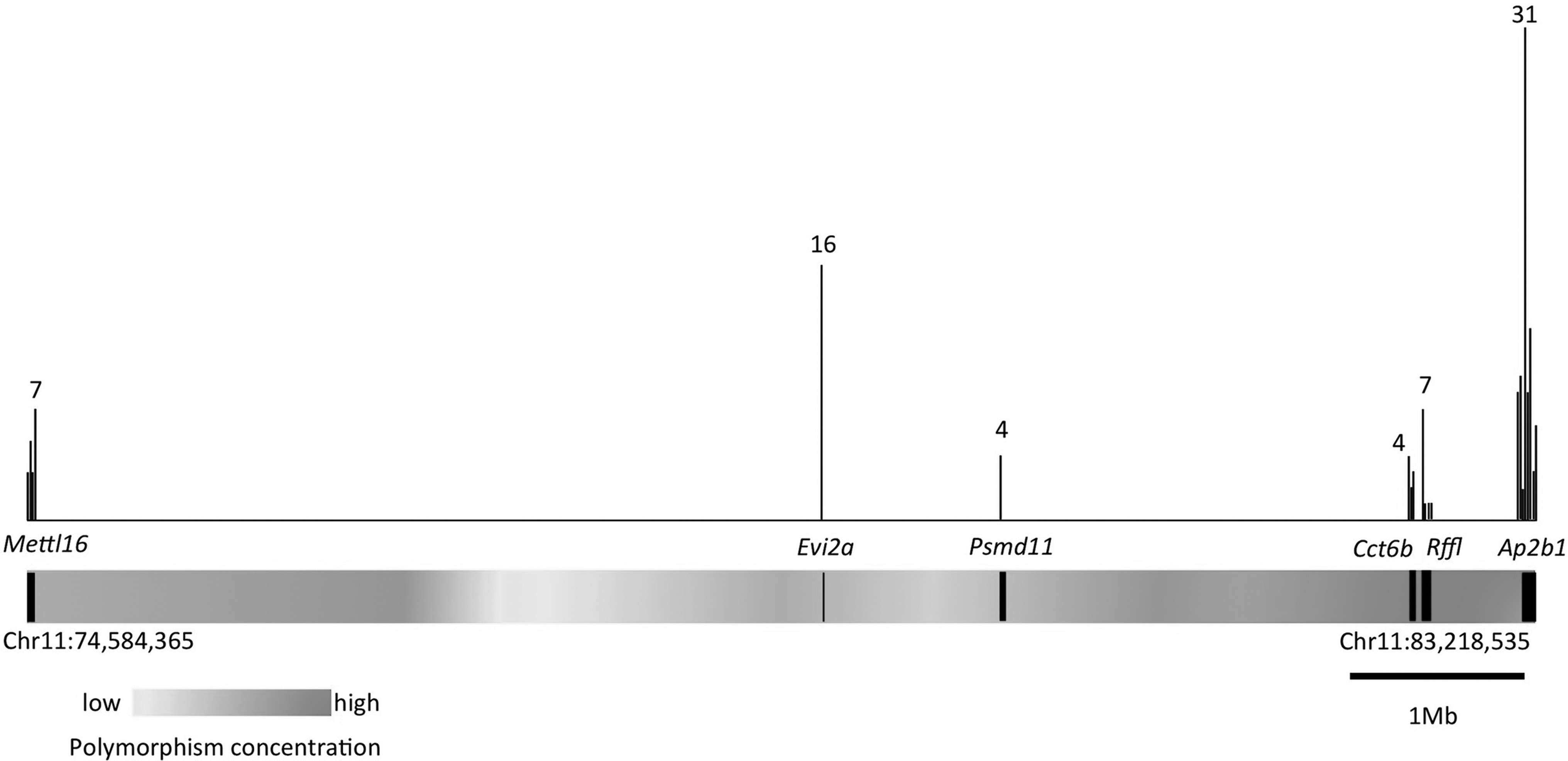
Density map of polymorphisms located in the domain of study. The heatmap represents current dbSNP information across this region. Dark gray indicates highest amount of SNPs and light gray stands for lowest amount. The six genes in this study are marked on the heatmap based on their chromosome locations and gene sizes. Genetic variations including SNPs and insertion/deletion polymorphisms identified in this study are shown by black lines. Each black line correlates with the number of polymorphisms within a 10 kb domain; longer lines signify increased polymorphism densities.
Discussion
The 6 genes in this study span a 6.8 Mb domain that contains a total of 154 genes, 9 miRNAs, and 5 snoRNAs. We concentrated our efforts on genes that are expressed during pre-implantation development or in gametes. The Evi2a locus, which is adjacent to Evi2b, resides within intron 33 of the Nf1 gene. Although the function of Evi2a is unknown, the locus demarcates the boundary between the two conserved regions, as it contains a high number of polymorphisms in a relatively small genomic region. We identified 16 genetic variants within the 4 kb Evi2a locus, whereas the average number of variants identified in this study is 6 within 10 kb. Although C57BL/6J, 129S6/SvEvTac, and 129S1/SvImJ are concordant for all variants of Evi2a, this gene could be crucial for the divergence of other mouse strains. Alternatively, polymorphisms within this intron could be important for Nf1 gene regulation. Consistent with this hypothesis, EVI2A is upregulated in malignant peripheral nerve sheath tumors in neurofibramatosis patients harboring microdeletions within the NF1 locus (Pasmant et al., 2011). The dbSNP database indicates that high levels of allelic variants are present in the distal end of the 6.8-Mb domain that overlaps with Cct6b, Rffl, and Ap2b1. This is consistent with our results, as we sequenced 84 variants within this locus.
The 129 strain is commonly used for knockout and other genetic manipulation studies because of the efficiency of obtaining embryonic stem (ES) cells that colonize to the germline (te Riele et al., 1992). This strain originated in 1928 and has diverged into more than 15 substrains. Different breeding strategies used in the past resulted in phenotypic and genetic diversity among these substrains (Simpson et al., 1997). In this report, we sequenced the 129S6/SvEvTac strain that has been maintained since 1992 at Taconic. According to Simpson et al. (1997), the origin of 129S6/SvEvTac can be traced back to the breeding of the Steel substrains, which resulted from the outcross of the 129/Sv strain to C3HeB/FeJ followed by 12–14 generations of backcrossing to the parental 129/Sv line. The resulting mice, termed 129/SvEv, were distributed to Martin Evans and were maintained by selection for the SteelJ allele. The 129/SvEv strains underwent further genetic manipulations to produce 129/SvEvBrd and 129/SvEv-Gpil c. Taconic crossed 129/SvEvBrd and 129/SvEv-Gpil c to generate 129S6/SvEvTac. This line has been bred as a separate inbred line at Taconic since 1992.
129S1/SvImJ was developed as a control-inbred strain for many of the Steel-derived strains (Petkov et al., 2004). Therefore, we included this strain as reference to compare the genetic variation between 129S1/SvImJ and 129S6/SvEvTac. Ap2b1, which has 84 polymorphisms across the locus, is the only gene in this study to contain variants between the 129S1/SvImJ and 129S6/SvEvTac inbred strains. There are 46 variants between C57BL/6J and 129S6/SvEvTac, 44 alterations between C57BL/6J and 129S1/SvImJ, and 5 SNPs between 129S6/SvEvTac and 129S1/SvImJ across the gene. Polymorphism number 33 (rs28210244) is a C1571G transversion in exon 11 that causes an F469L amino acid substitution. The remaining variants between the two 129 strains lie within introns or in the 3′UTR.
Ap2b1 encodes one of the two large chain components of the clathrin assembly protein complex 2. Ap2b1 protein is found on the intracellular domain of transmembrane-coated vesicles and is important for protein transport (Schmid et al., 2006). It is unclear why Ap2b1 contains so many polymorphisms and why there are differences between 129S6/SvEvTac and 129S1/SvImJ. It is possible that the genetic variations lead to alterations in gene regulation and/or protein structure changes that respond to strain-specific factors. However, it is equally likely that the high number of polymorphisms between 129S6/SvEvTac and 129S1/SvImJ is simply due to random silent mutations. Most of the variances within the Ap2b1 locus (82/84) lead to silent changes or are located within nonprotein-coding regions of the gene (i.e., introns or UTR), which is consistent with the latter explanation.
Although there are many variants between C57BL/6J and the 129-derived lines in this region of MMU 11, all of the polymorphisms between 129S6/SvEvTac and 129S1/SvImJ are located within a single gene. The domain proximal to Ap2b1 contains no variants between the two 129 strains. Regions of conservation suggest sequences critical for function, that is, gene variation is not functionally tolerated. Phylogenetic studies of nine different 129 strains indicates that the 129S6/SvEvTac and 129S1/SvImJ strains are highly related (Threadgill et al., 1997). Both were derived from a common ancestor bearing the Steel allele of the Kit ligand, although 129S1/SvImJ has been maintained at The Jackson Laboratories and 129S6/SvEvTac is maintained by Taconic. As six of the commonly used mouse ES cell lines have been derived from these two 129 strains, it is important to document the variants between these two 129 strains for future gene targeting and genotyping studies.
Conclusions
In this study, we sequenced 6 genes within a 6.8 Mb domain of mmu 11 and identified a total of 21 new SNPs and 6 Indels as well as confirmed genotype information for 114 additional SNPs among the C57BL/6J, 129S6/SvEvTac, and 129S1/SvImJ inbred strains. Most of these cluster at the two flanking loci, Mettl16 and Ap2b1. Ap2b1 is especially polymorphic among these three strains. These studies highlight the genetic variations among the three inbred strains and provide important information for understanding the evolutionary context of inbred strain divergence in mice.
Footnotes
Acknowledgments
The authors thank Lei Xing and Marianne Luu for excellent technical assistance, Drs. William Muir and Christopher Bidwell for critical reading of the manuscript, and Jason Fields for expert animal care. This project was funded by grants from the Purdue Research Foundation and generous start-up funds from the Department of Animal Sciences and the College of Agriculture at Purdue University.
Disclosure Statement
No competing financial interests exist.