
Abstract
The inflammatory response in prion diseases is dominated by microglia activation. The molecular mechanisms that lie behind this inflammatory process are not very well understood. In the present study, we examined the activat2ion of nuclear factor-kappa B (NF-κB) upon exposure to PrP106–126 and its role in PrP106–126-induced upregulation of inducible nitric oxide synthase (iNOS) and proinflammatory cytokines (interleukin [IL]-1β, tumor necrosis factor [TNF]-α, IL-6) in Ana-1 macrophages. The results showed that iNOS and proinflammatory cytokine release was significantly elevated in Ana-1 macrophages upon exposure to PrP106–126; that PrP106–126 treatment led to a significant NF-κB activation; that proinflammatory cytokines gene expression was elevated in macrophages upon exposure to PrP106–126; and that NF-κB inhibition significantly abrogated PrP106–126-induced upregulation of iNOS and inflammatory cytokine mRNA expression. These results suggest that treatment with neurotoxic prion peptides leads to the activation of transcription factor NF-κB, which in turn stimulates gene expression of iNOS and proinflammatory cytokines in Ana-1 macrophages.
Introduction
A synthetic peptide spanning human PrP region 106–126 (PrP106-126) recapitulates several chemicophysical characteristics of PrPSc (Tagliavini et al., 2001). PrP106-126 has been shown to elicit a diverse array of biological responses in mononuclear phagocytes (monocytes, macrophages, and microglia), including calcium mobilization, protein tyrosine phosphorylation, and cytokines production (Le et al., 2001). Mononuclear phagocytes appear to interact with the PrPC and are involved in prion diseases in many ways. PrPC can modulate phagocytosis in macrophages of wild-type and Prnp knockout mice, and this activity is important for normal macrophage functions and for the pathogenesis of prion diseases (Almeida et al., 2005; Zhou et al., 2009).
Nuclear factor-kappa B (NF-κB) is a transcriptional factor that plays a central role in inflammatory reactions (Whiteside et al., 1997). NF-κB is activated in response to a great number of stimuli, including the inflammatory cytokines interleukin-1 (IL-1), and tumor necrosis factor (TNF), hydrogen peroxide, ionizing radiation, viral infections, and bacterial lipopolysaccharide; most of these represent pathogenic stresses (Baeuerle, 1991). In peripheral tissues, the production of inflammatory mediators and acute phase proteins is mainly regulated by NF-κB, and NF-κB binding sites in their promoters serve as inducible transcriptional regulatory elements (Baeuerle and Henkel, 1994).
NF-κB pathway and proinflammatory cytokines that are capable of activating it have been previously linked to the pathogenesis of prion diseases (Fabrizi et al., 2001; Bacot et al., 2003; Zhou et al., 2008). In the current study, we characterized the ability of prion peptide PrP106-126 to activate NF-κB signaling pathways in mouse macrophage Ana-1 cells. Activated NF-κB was determined by western blot, and mRNA levels of inducible nitric oxide synthase (iNOS) and inflammatory cytokines IL-1β, TNF-α, and IL-6, were measured by quantitative real time-polymerase chain reaction (RT-PCR). The results indicate that PrP106-126 can activate NF-κB, and that this activation is involved in the expression of iNOS and inflammatory cytokines genes in PrP-induced murine Ana-1 macrophages.
Materials and Methods
Prion peptide preparation
PrP peptides PrP106–126 and scrambled PrP106–126 (Scr-PrP) with sequence of KTNMKHMAGAAAAGAVVGGLG and AVGMHAGKGLANTAKAGAMVG were synthesized by Sangon Bio-Tech. The peptides were dissolved in Phosphate Buffer Solution (PBS) at a concentration of 2 mM and stored at −20°C as the stock solutions. The purity of prion peptides was >95% according to the synthesizer's data.
Reagents and antibodies
NF-κB inhibitor (BAY 11-7082) and antibodies against kBa were obtained from Beyotime Biotechnology, Inc.
Cell line and culture
Ana-1 cells, a murine macrophage cell line, were maintained in a 95% air, 5% CO2 atmosphere in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Gibco), 100 μg/mL streptomycin, 100 U/mL penicillin (Gibco), and 2 mM glutamine per milliliter.
Peptide treatment
Prion peptide 106–126 was aggregated for 72 h at room temperature in RPMI 1640 medium to increase fibrillogenicity of the peptide. Ana-1 cells were exposed to 12 μM NF-κB inhibitor for 1 h and then treated with the aggregated peptide PrP106–126 in culture medium. ScrPrP was used as a negative control. Three wells were used in each group of experimental conditions.
Nuclear extract and western blot
After being treated with 25 μM PrP106–126 for 24 h, the culture medium was discarded, and nuclei proteins were extracted using Nuclear Extract kit (Beyotime Biotechnology, Inc.). Equal amounts of protein were separated by sodium dodecyl sulfate - polyacrylamide gel electrophoresis on 8% gels, and the separated proteins were transferred onto a nitrocellulose membrane. Nonspecific binding sites were blocked by incubating the membrane with 5% fat-free dried milk in Tris-Buffered Saline Tween-20 (TBST) (10Mm TrisHC1, pH 7.5, 0.15M NaCl, 0.05% Tween20). A mouse anti-β-actin monoclonal antibody (1:1000) or a rabbit anti-p65 polyclonal antibody (1:1000) was added and incubated for 1h at 4°C. Membranes were washed with TBST and then incubated with the secondary antibody, an anti-mouse or anti-rabbit horseradish peroxidase-conjugated goat antiserum (1:5000). Bands of immunoreactive protein were visualized, after membrane incubation with enhanced chemiluminescence (ECL) reagent for 5 min, on a Bio-Rad image system. The blot was stripped and reprobed with anti-actin antibody to estimate the total amount of protein loaded.
Cytokines release assessment
The levels of interleukin-1β (IL-1β), IL-6, and TNF-α were determined in culture supernatants of Ana-1 cells (0.2×106 cells/cm2) treated for 24 h with the peptides (50 μM) by using enzyme-linked immunosorbent assay (ELISA) kits specific for mouse IL-1β, IL-6, and TNF-α. The samples of culture supernatants, controls, and standards were first treated by Fast Protein Precipitation and Concentration KIT (Wuhan Boster Biotech) to increase protein concentration before being used for ELISA analysis. Samples were then pipetted into microplates of these ELISA kits, according to the manufacturer's instructions (R&D Systems). The experiments were performed in duplicate, in four to six independent cell preparations. The absorbance was measured at 450 nm, with the correction wavelength at 540 nm, using microplate reader. The values were read off the standard curve and expressed as nanograms per liter (IL-1β, IL-6, TNF-α) or U/L (i-NOS).
Immunofluorescence
Cells were pretreated or not with BAY 11-7082, exposed to PrP 106–126, fixed in 4% paraformaldehyde (15 min at room temperature), rinsed with PBS, blocked with 1% bovine serum albumin /PBS (30 min at room temperature), and then incubated with the primary antibody (Rabbit anti-p65 diluted 1:300 in blocking solution) for 1 h at room temperature. Cells were then rinsed with PBS, incubated with goat anti-rabbit IgG (H+L) diluted 1:500 in blocking solution for 1 h at room temperature. Cells were then rinsed with PBS and viewed on an Olympus FV500 laser confocal scanning system.
Total RNA isolation
Ana-1 cells were stimulated by prion peptide 106–126 using the same treatment conditions as above, and RNA-Solve reagent (Omega Bio-tek) was added. The total RNA of macrophages was extracted according to the manufacturer's instruction. The RNA extracts were treated with RNase-free Dnase I to remove DNA, and quantified on a spectrophotometer (BioPhotometerw Eppendorf), then stored at −80°C.
Quantitative RT-PCR of cytokines
The RNA from each sample was reverse transcribed with oligodT to cDNA using Reverse Transcription System (Promega). To determine the mRNA expression of cytokines, real-time PCR was carried out using DNA Engine OpticonTM 2 continuous fluorescence detection system and SYBR Green PCR Master Mix kit (Tiangen Biotech). Endogenous house-keeping gene β-actin was used as a cDNA template control. PCR primers were used for amplification of genes cloned for real-time PCR (see Table 1). Each PCR reaction contained 500 nM of each primer, 2 μL cDNA, 10 μL Power SYBR Green PCR Master Mix (2X) in a final volume of 20 μL. After an initial denaturation for 10 min at 95°C, the PCR reaction was 40 cycles of amplification consisting of denaturation at 94°C for 15 s, followed by annealing step at 60°C for 20 s, and extension at 72°C for 20 s. A final elongation step at 72°C for 8 min concluded the PCR. The reaction was then subjected to a melting protocol from 65°C to 95°C with a 0.2°C/s increment and 1 s holding at each increment to examine the specificity of the amplified products. Data were collected using PCR baseline deduction mode. After adjusting baseline cycles and calculating threshold value, the sample threshold cycle was obtained. All data were analyzed by software SPSS13.0. An independent sample t-test was used to analyze differences in mRNA expression between different groups. Differences with p<0.05 were considered to be statistically significant. All samples were analyzed in triplicate.
PCR, polymerase chain reaction; IL, interleukin; TNF, tumor necrosis factor.
Statistical analysis of quantitative PCR
Comparison of treatment effects was carried out using one-way analysis of variance techniques of SPSS 13.0. Data are expressed as mean±SD. Differences with p<0.05 were considered statistically significant.
Results
PrP106-126 induced an increase in protein levels of proinflammatory cytokines in Ana-1 cells
PrP106-126 treatment increased iNOS and proinflammatory cytokines IL-1β, TNF-α, and IL-6 release, and there was a statistically significant difference. The protein levels of the four cytokines were higher in PrP106-126-treated cells compared with ScrPrP- or PBS (control)-treated cells (p<0.05) (Fig. 1).
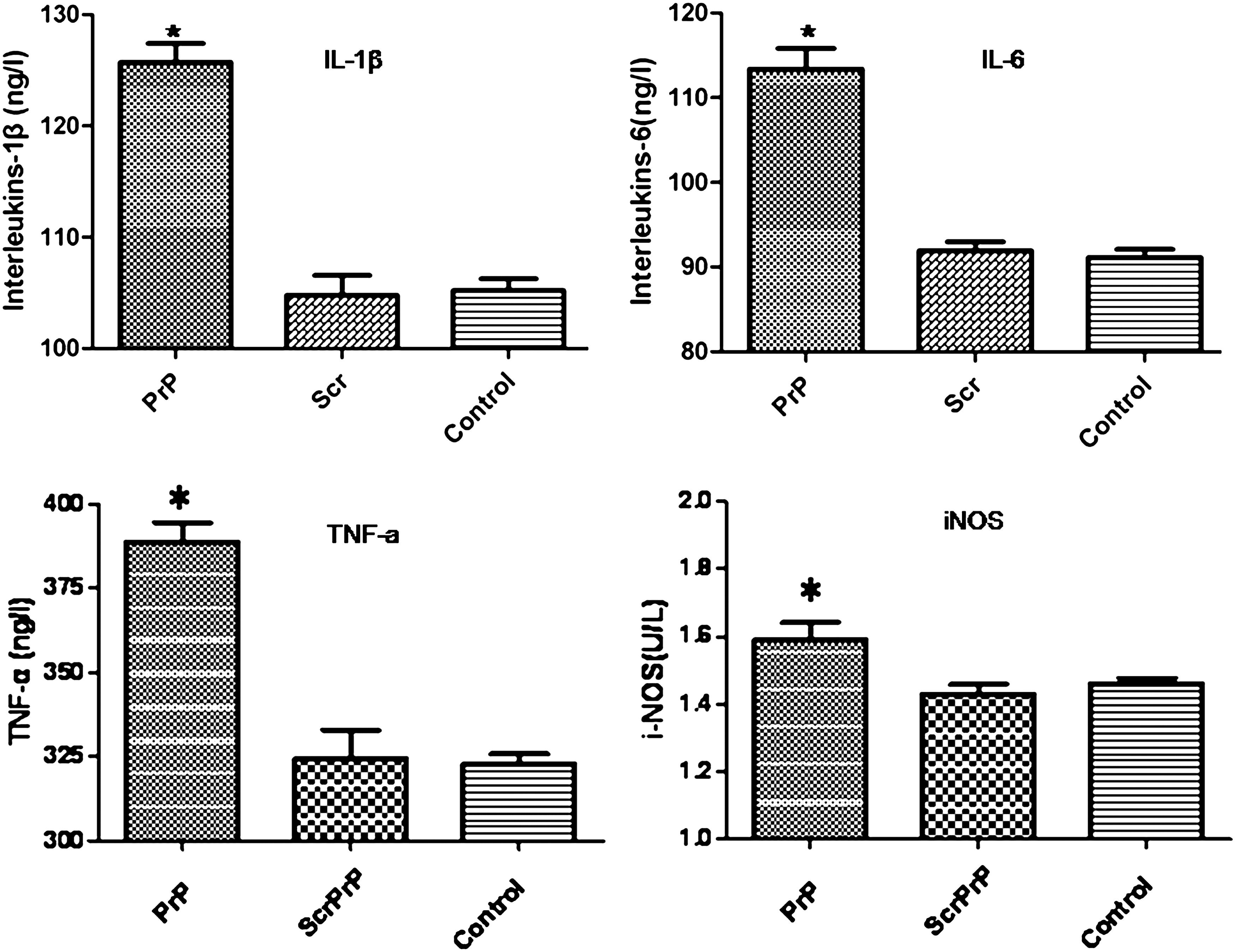
Cytokines produced by Ana-1 cells treated with PrP106–126. Cells were treated for 24 h with PBS, ScrPrP (25 μM), or PrP106-126 (25 μM). The levels of proinflammatory cytokines were determined in culture supernatants by ELISA as described in Materials and Methods. All data are mean±SD of triplicate samples and are representative of 3 experiments. *p<0.05. ELISA, enzyme-linked immunosorbent assay; SD, standard deviation; PBS, Phosphate Buffer Solution; IL, interleukin; TNF, tumor necrosis factor; iNOS, inducible nitric oxide synthase.
PrP106-126 induced NF-κB activation in Ana-1 cells
To investigate the role of NF-κB in PrP106-126-induced increase in cytokine release in Ana-1 macrophages, we first investigated the effect of PrP106-126 treatment on NF-κB activation through examining the nuclear translocation of p65, a component of NF-κB, by western blot analysis. Cytoplasmic and nuclear extracts were prepared from Ana-1 macrophages treated with PrP106-126, ScrPrP, or PBS. As shown in Figure 2, 24 h-treatment with 25 μM PrP106-126 induced significant nuclear translocation of NF-κB p65 subunit (PrP) from cytoplasm to cytoblast accompanied by the decrease in the level of cytoplasmic NF-κB p65. The relative density of translocated p65 band in the cytoblast was significantly higher in cells treated with PrP106-126 than in those treated with ScrPrP or PBS (control).
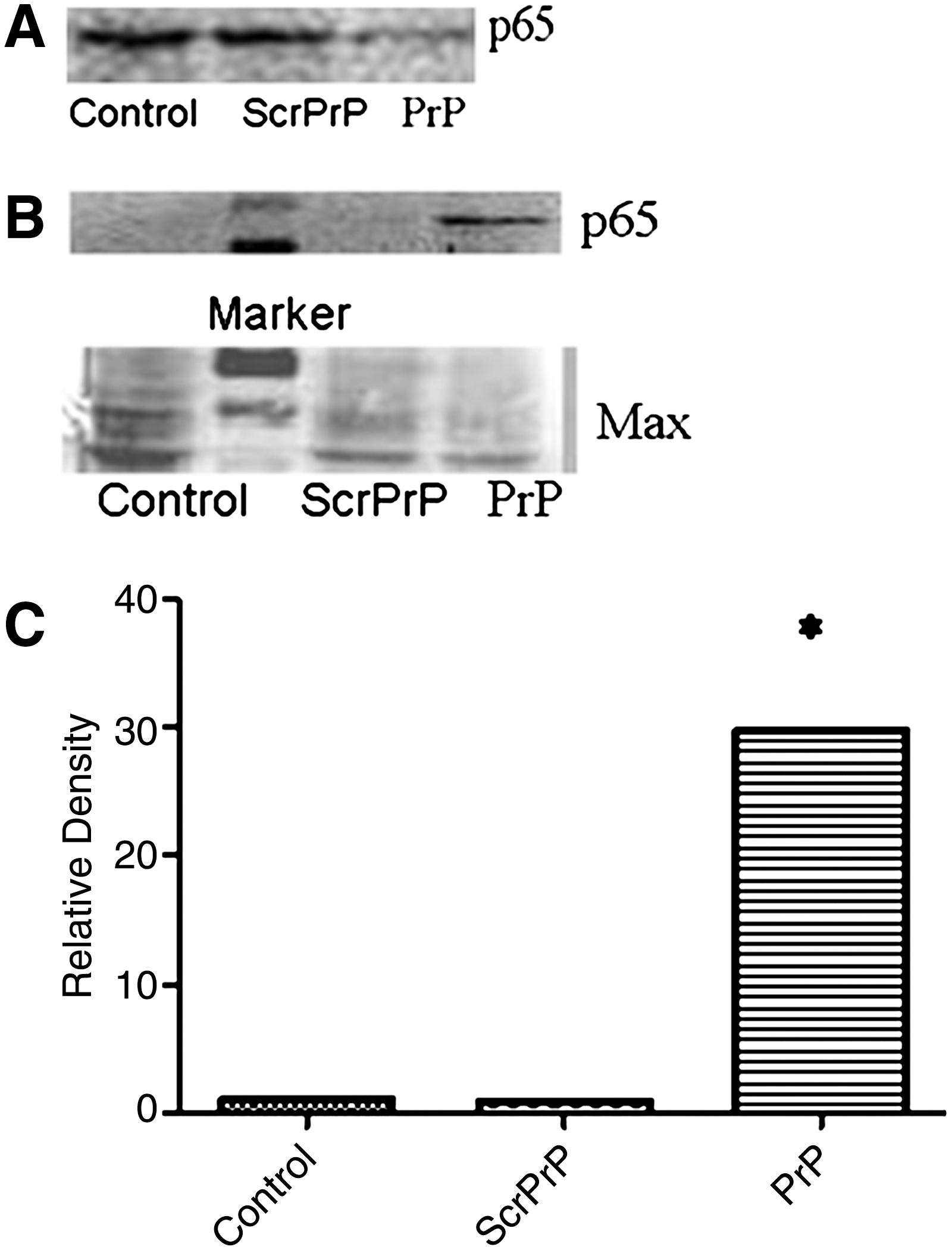
Western blot analysis of p65 nuclear translocation. Cells were treated with 25 μM PrP106-126, 25 μM ScrPrP, or PBS.
PrP106-126 induced NF-κB-dependent increase in the mRNA levels of proinflammatory cytokines in Ana-1 cells
To investigate the involvement of NF-κB activation in PrP106-126-induced increase in inflammatory cytokine release, changes in proinflammatory cytokines and iNOS mRNA levels were quantified in PrP106-126-treated cells preincubated or not with NF-κB inhibitor BAY 11-7082.
Ana-1 cells were treated with 25 μM PrP106-126 or ScrPrP for 24 h. IL-1β, TNF-α, IL-6, iNOS, and β-actin genes were amplified and confirmed by gel electrophoresis and gene sequencing (Fig. 3). PrP106-126 treatment upregulated the mRNA expression of iNOS and the proinflammatory cytokines IL-1β, TNF-α, and IL-6 (Fig. 4). Pretreatment with NF-κB inhibitor on the Ana-1 cells inhibited the increase of IL-1β, TNF-α, IL-6, and iNOS mRNA level induced by PrP106-126 (p<0.05) (Fig. 4). The mRNA level of TNF-α, IL-1β, IL-6, and iNOS were much higher in treated cells compared with those pretreated with NF-κB inhibitor and ScrPrP- or PBS-treated cells (p<0.05) (Fig. 4).
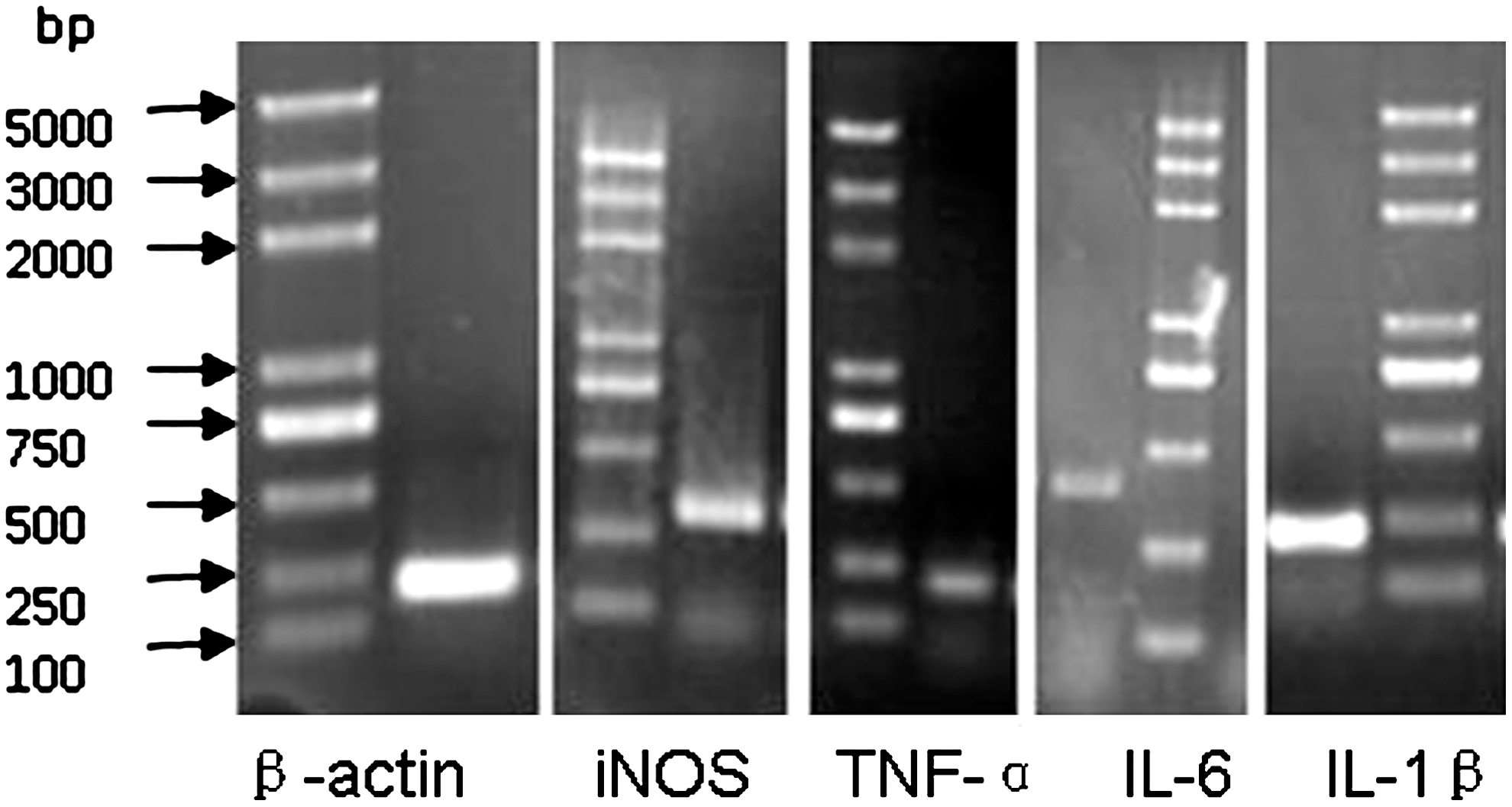
Electrophoresis of RT-PCR-amplified proinflammatory cytokine and iNOS genes on an 8% agarose gel stained with ethidium bromide. β-actin, 223 bp; iNOS, 312 bp; TNF-α, 221 bp; IL-6, 384 bp; IL-1β, 229 bp. RT-PCR, real time-polymerase chain reaction.

The mRNA expression of iNOS and proinflammatory cytokines in Ana-1 cells. Cells were preincubated or not with NF-κB inhibitor BAY 11-7082, and then treated with 25 μM PrP106-126 or ScrPrP for 24 h. The mRNA levels of iNOS and inflammatory cytokines IL-1β, TNF-α, and IL-6 were measured by quantitative RT-PCR. The expression level was shown as relative expression in different groups. PrP: Cells were treated with PrP106-126; iPrP: Cells were preincubated to the NF-κB inhibitor (12 μM) for 1 h before being treated with PrP106-126; Scr: Cells were treated with ScrPrP; iControl: Cells were preincubated with the NF-κB inhibitor for 1 h and were treated with PBS; Control: Cells were treated with PBS. Results were expressed as the mean±SD. *p<0.05. NF, nuclear factor.
The effect of NF-κB inhibitor was confirmed by immunofluorescence analysis and western blot analysis of NF-κB nuclear translocation in cells treated or not with NF-κB inhibitor, the result shows that the NF-κB activated by PrP106-126 and translocated from cytoplasm to cytoblast. The analysis of relative density revealed that there was a statistically difference between the cells treated and untreated with NF-κB inhibitor (Fig. 5).
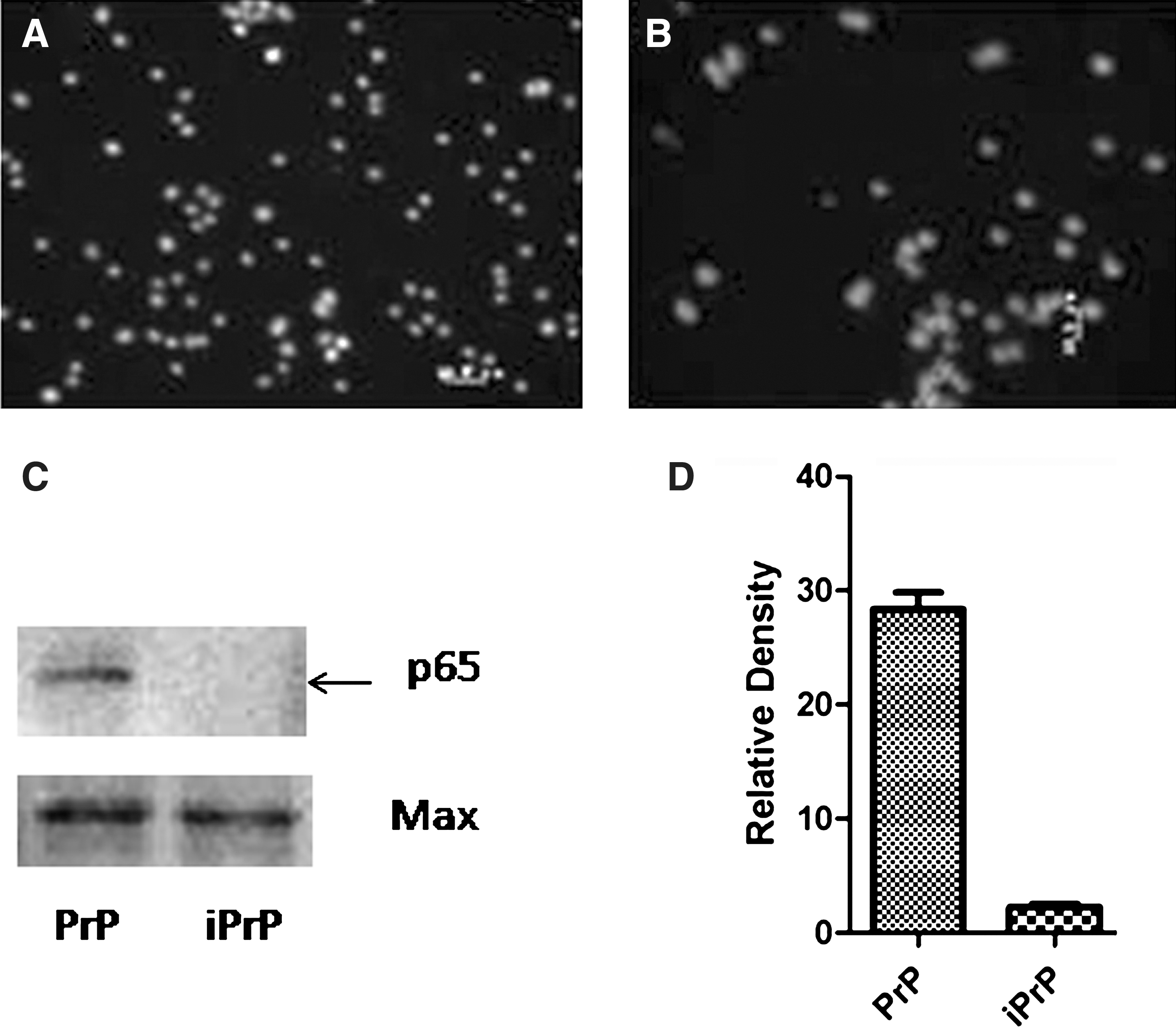
Inhibition of NF-κB by BAY 11-7082 in Ana-1 Cells. Cells were preincubated or not with BAY 11-7082, and then exposed to PrP106-126. The inhibitory effect of BAY 11-7082 was confirmed by examining nuclear translocation of NF-κB using immunofluorescence analysis and western blot analysis.
Discussion
NF-κB system is a major regulator of innate and adaptive immunity and inflammatory responses (Baeuerle and Henkel, 1994; Doyle and O'Neill, 2006; Perkins, 2007). NF-κB activation has been linked to gene regulation in cellular processes such as inflammation, immune response, cell proliferation, and apoptosis (Chen et al., 2002). The NF-κB transcription factor chiefly resides in the cytoplasm, where it is located as an inactive complex bound to IκB-α. Upon extracellular stimulation, IκB-αs is phosphorylated and subsequently degraded, and that leads to the release of NF-κB, the nuclear translocation p65 subunit of NF-κB, and the activation of target gene expression (Baeuerle and Baltimore, 1996; Bai, 2008).
Inflammation is an important component in the pathogenesis of several age-related degenerative diseases (Salminen et al., 2008). Induction of IL-6 and iNOS gene expression is mediated by NF-κB activation in IL-1β and Aβ-treated astroglial cultures (Chao et al., 1997; Forloni et al., 1997; Akama et al., 1998; Bales et al., 1998).
In the present study, we reported that NF-κB signaling plays a central role in PrP106-126-induced iNOS and proinflammatory cytokine gene upregulation at mRNA level in Ana-1 cells. We demonstrated that NF-κB was activated in Ana-1 macrophages upon exposure to PrP106-126, and that NF-κB activation led to the increase of proinflammatory cytokine gene expression, since pretreatment with NF-κB inhibitor significantly abrogated PrP-induced upregulation of proinflammatory cytokine gene expression.Taken together, these results indicate that PrP106-126 induced upregulation of inflammatory cytokine genes is NF-κB dependent.
These results are consistent with other reported studies related to neurological disorders including TSE, Parkinson's disease, and Alzheimer's disease (Kim et al., 1999; Bai, 2008; Li et al., 2008; Yang, 2008; Zhou et al., 2008), and elucidate some of the molecular mechanisms involved in the interaction between PrP and immune cells. However, they do not exclude the possibility that other signaling pathways might be involved in the stimulation of proinflammatory cytokines gene expression during macrophage-prion interactions. Although further studies are needed to investigate the role of other possible signaling pathways in the interaction between PrP and immune cells, our study contributes to the understanding of the molecular mechanisms lying behind the interaction between PrP and macrophages.
Footnotes
Acknowledgments
This work was supported by the Ministry of Agriculture, Key Program, China (Project No. 2009ZX08008-010B), the Ministry of Agriculture Key Program, China (Project No.2009ZX08007-008B), the Natural Science Foundation of China. (Project No. 31001048, No. 30972164, and No. 30871854), and the Program for Cheung Kong Scholars and Innovative Research Team in the University of China (No. IRT0866).
Disclosure Statement
No competing financial interests exist.