
Abstract
Hypermethylated genomic DNA is a common feature in tumoral tissues, although the prevalence of this modification remains poorly understood. We aimed to determine the frequency of five tumor suppressor (TS) genes in prostate cancer and the correlation between promoter hypermethylation of these genes and low and high grade of prostate carcinomas. A total of 30 prostate tumor specimens were investigated for promoter methylation status of TS hypermethylated in cancer 1 (HIC1), death-associated protein kinase 1 (DAPK1), secreted frizzled-related protein 2 (SFRP2), cyclin-dependent kinase inhibitor 2A (p16), and O-6-methylguanine-DNA methyltransferase (MGMT) genes by using bisulfite modifying method. A high frequency of promoter hypermethylation was found in HIC1 (70.9%), SFRP2 (58.3%), and DAPK1 (33.3%) genes in tumor samples that were examined. The current data show high frequency of hypermethylation changes in HIC1, SFRP2, and DAPK1 genes in prostate carcinomas of high Gleason Score (GS).
Introduction
In the current study promoter hypermethylation of 5 TS genes; p16INK4a, hypermethylated in cancer 1 (HIC1), death-associated protein kinase 1 (DAPK1), secreted frizzled-related protein 2 (SFRP2), and O-6-methylguanine-DNA methyltransferase (MGMT) were investigated in prostate cancer patients with different Gleason Score (GS). The p16INK4a gene, a cyclin-dependent kinase inhibitor and founding member of INK4 family, inhibits the association between cyclin-dependent protein kinase 4/6 (CDK4/6) and D-type cyclins, and blocks phosphorylation of retinoblastoma 1 (RB) (Chin et al., 1998). DAPK1 is a multi-domain, calmodulin -regulated serine threonine protein kinase that mediates a range of processes, including apoptosis, autophagy, and tumor invasion (Eldik, 2002; Kuo et al., 2006; Lin et al., 2010). SFRP2 is one of the negative modulators of Wnt-frizzled signal transduction pathway which plays an important role in normal development and oncogenesis (Mii and Taira, 2009). MGMT is a DNA repair enzyme which catalyses the transfer of alkylating agents (mainly a methyl group from O6-methylguanine) in DNA (Zhong et al., 2010). TS HIC1 is an essential gene for mammalian development (Carter et al., 2000) and epigenetically inactivated in some types of human cancer (Wales et al., 1995). Hypermethylation mostly lies between the intron 2 and exon 3 of HIC1 gene and suppresses age-dependent development of cancer by inactivating p53 (Chen et al., 2005). The gene, located on the distal arm of chromosome 17p13.3, encodes a sequence specific zinc-finger transcriptional factor (Pinte et al., 2004) and has a consensus p53 protein binding site in its promoter (Wales et al., 1995; Britschgi et al., 2006). HIC1 is transcribed from two promoters and dense methylation of either HIC1 promoter is associated with complete loss of transcription (Chen et al., 2005; Fleuriel et al., 2009). High density of methylation HIC1 promoter is associated with aggressiveness of the tumor and poor overall survival (Wales et al., 1995; Nicoll et al., 2001; Hayashi et al., 2001; Rood et al., 2002; Waha et al., 2004).
Structural genetic changes have been widely characterized in prostate carcinoma, while epigenetic changes are poorly reported. We aimed to evaluate the frequency of CpG-island hypermethylation in some cancer-associated TS gene promoters in different grades of prostate tumors which may have potential as a diagnostic and/or prognostic biomarker in the presented results.
Methods
Patient and tumor specimens
This study was approved by the Research Ethics Committees of Faculty of Medicine of Cumhuriyet University. Fresh tumoral tissue samples from 30 patients who underwent transurethral resection and open radical retro pubic prostatectomy in the Department of Urology between January 2006 and December 2010 were used in the current study.
Patient's mean age was 70.23 (51–85) years. Digital rectal examination (DRE) was normal in 12 patients whereas the rest were abnormal. Mean prostate-specific antigen (PSA) value was 71.76 ng/dL. Transurethral resection of prostate was performed in 22 of 30 patients because they had a normal PSA level and normal DRE or they had lower urinary tract symptoms associated with advanced disease or other co-morbidities. Radical retro pubic prostatectomy was performed in 7 of 30 patients. In one patient who underwent radical cystoprostatectomy with muscle invasive bladder carcinoma, pathologic examination revealed prostate cancer. Histopathologic evaluation revealed adenocarcinoma in 29 patients and small cell prostate cancer in 1 patient. Five patients had low grade cancer (GS ≤6) and 24 patients had high grade cancer (GS ≥7–10). The number of patients who had local disease were 8 and 22 of 30 patients had advanced disease.
Methylation specific-polymerase chain reaction and DNA converting
Sodium bisulfite mediated methylation specific-polymerase chain reaction (MSPCR) technique was used to determine the epigentical alterations in the tumoral tissues. This technique deaminates cytosine base in DNA, while 5-methylcytosine resists this action (Boyd and Zon, 2004; Hayatsu, 2008). MSPCR is a sensitive method that discriminately amplifies and detects a methylated region of interest by using methylation site-specific primers on bisulfite-converted genomic DNA. This deamination leads to the conversion of cytosines into uracil residues, which are recognized as thymines in subsequent PCR amplification, whereas the modified cytosines do not react and are therefore detected as cytosines (Clark et al., 2006). Such primers will only anneal to sequences that are methylated and thus contain 5-methylcytosines that are resistant to conversion by bisulfite. Modified and amplified DNA fragments are detected by reverse hibridization of PCR products with commercially available test strips.
Analysis of methylation status of the promoter region of TS genes
Tumoral samples were histologically graded based on the WHO/ISUP and staged according to the TNM classification and examined for tissue specific epigenetic alterations. All fresh specimens were used for detecting possible epigenetic modifications in TS genes. After total genomic DNA isolation genes were modified by sodium bisulfite modification, methylation patterns were determined and correlated with standard clinico-histopathological parameters. DNA methylation patterns in the promoter CpG islands were determined in tumoral tissue samples by MSPCR following the bisulfite modification technique. Forty-five microliters of isolated DNA were denatured by alkalizer (final volume 0.3 mM) at 55°C for 10 min and modified by sodium bisulfite (5.20–5.69 M, pH 5.0; Viennalab) for 4 h at 55°C in the dark. After incubation, binding buffer was added (300 μL per sample) and lysate was transferred into a receiver tube with spin filter, centrifuged at 13 000 rpm for 30 s. Filtrate was discarded, wash buffer (600 μL per sample) was added into the spin filter and centrifuged at 13 000 rpm for 30 s. A mixture of alkalizer ethanol (1:10) was added into the spin filter, 300 μL per sample, then incubated at room temperature for 30 min. After incubation, spin filters were washed again. Elution buffer (30 μL) was added into spin filters, incubated 3 min at room temperature, centrifuged at 13 000 rpm for 1 min, and filters discarded. The resulting filtrate was keep at −20°C. Aliquots of 5 μL of bisulfite modified DNA were used for MSP reactions. Primers for a methylated and unmethylated promoter of the target TS genes (SFRP2, p16, DAPK1, HIC1, and MGMT) were used and multiplex PCR based amplification procedure (Viennalab) was performed for in vitro gene amplification. Multiple PCR products were carried out in a Bioer XP thermal cycler after 15 min at 94°C for Pre-PCR, 45 s at 94°C for denaturation, 45 s at 66°C for annealing, and 45 s at 72°C for polymerization of 45 cycles with final extension for 7 min at 72°C. PCR products that amplified from modified DNA from tumoral tissue (10 μL of each PCR reaction) were compared in reverse hybridization strip assay technique (ViennaLab), (Ozdemir et al., 2010). One signal was evaluated as heterozygous inactive (inactivation of one allele); double signals were homozygous hypermethylated (inactivation of both alleles), and samples without any signals were evaluated as hypomethylated (fully active gene). For statistical evaluation, the SPSS 14.0 for Windows was used. The parameters of the patients are presented as mean±standard deviation and p<0.05 was considered statistically significant.
Results
By MSPCR technique, we evaluated the methylation status for HIC1, SFRP2, DAPK1, and MGMT TS genes in prostate tumors with different GSs. We found that HIC1 were hypermethylated in 70.1% (17/24) in prostate adenocarcinomas with high GSs.
Clinicopathologic data and follow-up knowledge
A total of 30 male patients with mean age 70.23 with prostate carcinomas were clinically diagnosed and treated. Solid tumoral tissues (1 small cell prostate carcinoma [SCPC] and 29 adenocarcinoma) were examined in the current study. The number of patients investigated according to stages of tumors were; 12 in pT1, 8 in pT2, and 10 in pT4. GS was between 6 and 8 in patients with pT1, 8 in patients with pT2, and 9 or 10 in all patients with pT4 tumors (Table 1).
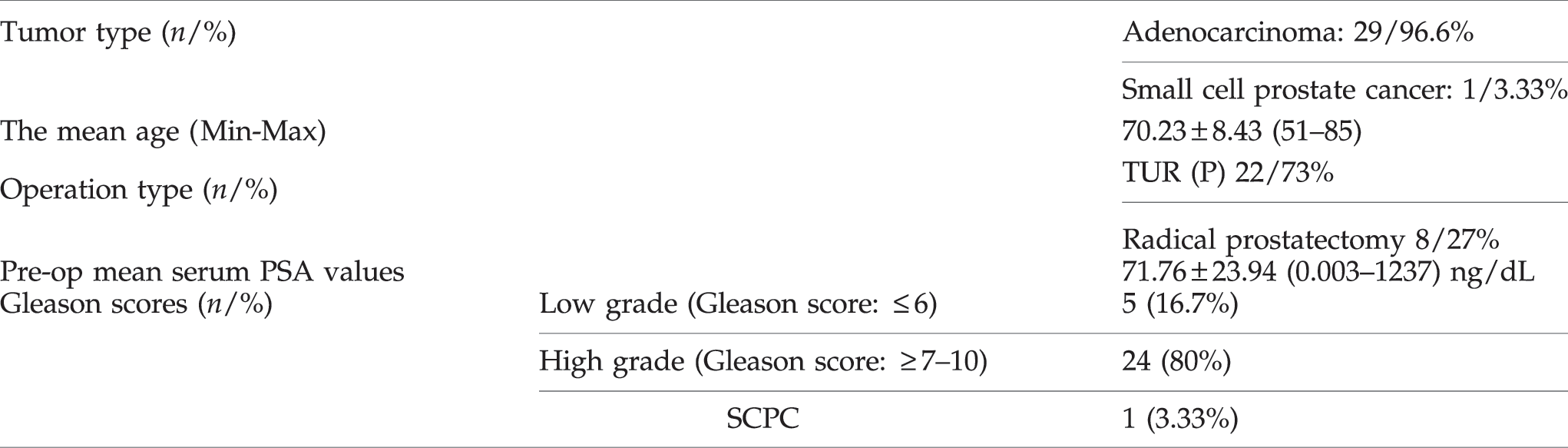
PSA, prostate-specific antigen; SCPC, small cell prostate carcinoma.
Increased percentage of promoter hypermethylation in HIC1, SFRP2, and DAPK1 genes and the clinical and histopathological parameters in prostate cancer of 30 samples showed a high sensitivity and specificity. There was no promoter hypermethylation-gene inactivation in TS p16 in all tumor samples examined (100% of tumors were hypomethylated). Tumor specimens showed high frequency of promoter hypermethylation in HIC1 (70.9%), SFRP2 (58.3%), and DAPK1 (33.3%) genes.
The HIC1 gene was fully inactive (hypermethylated) in one case of SCPC but the rest of studied genes were fully active (100%) in that case (Table 2). In general, various (heterogeneous type) epigenetic alterations were detected for some TS genes in distinct prostatic adenocarcinomas of different GS (Tables 1 and 2). TS DAPK1 (33.3%) and MGMT (8.3%) showed partial and/or full inactivation in early and/or late stage tumors (Table 2), (Fig. 1). High frequency of full promoter hypermethylation (homozygous for both alleles) for HIC1 (70.9%) and SFRP2 (58.3%) genes were detected in prostate tumor specimens that were examined. Two groups of tumors with different GS (Group 1, GS ≤6 and Group2, GS ≥7) were statistically compared. While HIC1 hypermethylation profiles were significant for both groups, only SFRP2 gene showed statistical significance for GS ≥7 in the current tumoral tissues (p>0.016, Table 2). No difference was found between GS for the rest of the genes that were examined (p>0.016, Table 2).
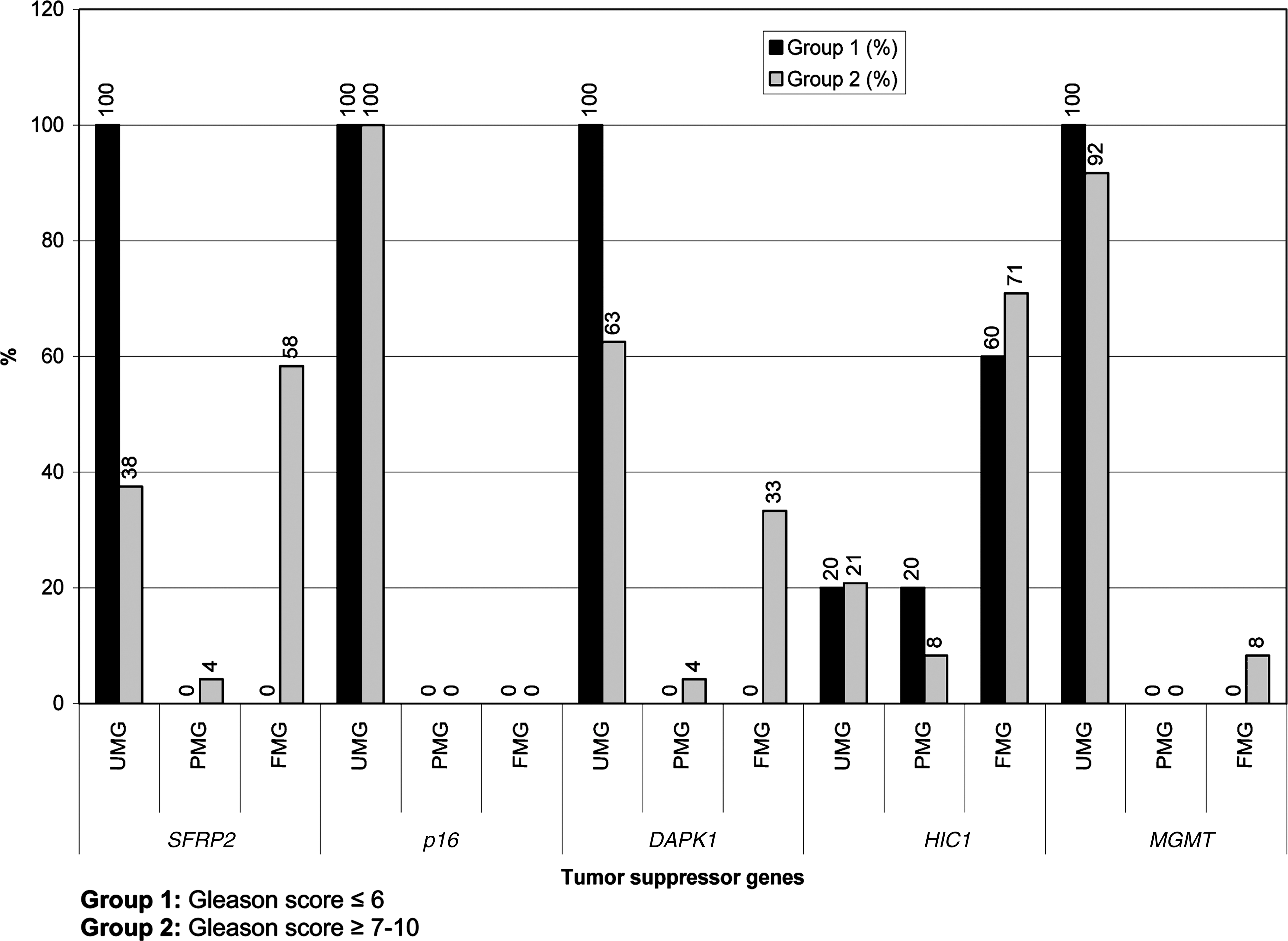
Bar graphs show the methylation ratios of target tumor suppressors studied in the current patients. UMG, unmethylated gene; PMG, partially methylated gene; FMG, fully methylated gene.
GS, Gleason score.
Discussion
Promoter hypermethylation which repress transcription of the TS genes leading to gene silencing has been extensively studied. Epigenetic alterations can disrupt TS gene function as an alternative to inactivating genetic mutations (Jones and Baylin 2002; Brophy et al., 2003; Zhang et al., 2006; Marsit et al., 2006a; Schulz and Hofmann, 2009). Recent developments in epigenetic methodologies and gene silencing techniques opened a new area for the identification of epigenetic parameters (Marsit et al., 2006b; Nojima et al., 2007; Shih et al., 2007; Hoffmann et al., 2007; Awakura et al., 2008). Although the mechanisms of specific gene hypermethylation in cancer precursor cells are unknown, locus-specific de novo hypermethylation may occur in those cells by hyperactivity of DNA methyltransferase enzyme as claimed by Simon and Lange (2008).
In the present study, we determined the hypermethylated profiles for promoter regions of target TS genes HIC1, p16, MTMG, DAPK1, and SFRP2 in prostate cancers. SFRP1 is a marker of higher tumor stage, grade, and poorer survival in renal cell carcinoma (Dahl et al., 2007). Marsit et al., (2005) have reported methylation-induced epigenetic alterations in SFRP genes associated with the occurrence and/or progression of bladder cancer. In our previous study, full inactivation of SFRP2 was reported in two cases of fistula associated mucinous type anal adenocarcinomas (Sen et al., 2010). SFRPn proteins are also responsible for the constitutive inactivation of WNT signaling (Wolff et al., 2005). Hypoexpression due to hypermethylated SFRP2 was reported in 94.2%, 52.4%, 37.5%, and 16.7% of patients with colorectal carcinoma (CRC), adenocarcinomas, hyperplastic polyps, and ulcerative colitis, respectively (Huang et al., 2007). HIC1 encodes a zinc-finger transcription factor that is essential for mammalian development (Esteller, 2000). HIC1 forms a transcriptional repression complex with NAD-dependent deacetylase sirtuin-1 (SIRT1) deacetylase, and this complex directly binds the SIRT1 promoter and represses its transcription. The inactivated form was reported in a few types of human cancer (Berezovska et al., 2006) and is epigenetically inactivated, but not mutated, in some human cancers (Van Leenders et al., 2007). Inactivation of HIC1 results in upregulated SIRT1 expression in normal or cancer cells; this deacetylates and inactivates p53, allowing cells to bypass apoptosis and increases cancer risk in mammals.
In the current study we aimed to find hypermethylation frequency of the 5 target TS genes and to correlate this data with clinical findings. Results show high frequency of promoter CpG methylation of various TS genes (HIC1, SFRP2, and DAPK1) in prostate tumors and their association with high Gleason grade. These data might suggest a role for HIC1 in prostate carcinogenesis and indicate that HIC1 promoter methylation might be a diagnostic and prognostic biomarker in prostate cancer. In the low GS group (GS ≤6; n=5) only HIC1 was hypermethylated (n=1, 20% partially methylated and n=3, 60% fully methylated). In the high GS group (GS ≥7; no=24) the HIC1 gene was also hypermethylated (n=2, 8.3% partially and n=17, 70.9 fully hypermethylated). Morton et al. (1996) have showed that the hypermethylation of chromosome 17p locus D17S5 is a tissue-specific event in prostate DNA, and they hypothesized that methylation of this and/or related loci may play a role in the extreme predilection of this gland to neoplastic growth. Only the SFRP2 gene showed statistically significant relationship with GS ≥7 tumors (p>0.016, Table 2). Different frequency of hypermethylation profiles were detected for the other TS genes in both groups but the results were not statistically significant (Table 2). Results confirmed the association between gene hypermethylation status and advance tumoral differentiations. The current results were also supported by other findings (Catto et al., 2005; Kim and Kim, 2009). They found that promoter hypermethylation was present in 86% of transitional cell carcinoma and also occurred more frequently and more extensively in urinary tract tumors (94%) than in bladder tumors (Catto et al., 2009). No correlation was found between p16, MGMT gene inactivation and tumor grade, stage, recurrence, progression and/or invasion. Tumoral specimens showed fully methylated profiles for HIC1 (70.9%), SFRP2 (58.3%), and a partially hypermethylated profile for the DAPK1 (33.3%) and MGMT (8.3%) genes in different ratios according to the tumor grade (Table 2, Fig. 1). The current data also shows a high frequency of hypermethylation changes in the HIC1 and SFRP2 TS genes. DAPK1 gene, a positive regulator for apoptosis, showed promoter hypermethylation frequency at 33.3% for the high GS group in the prostate cancer samples.
HIC1 is a TS gene which is epigenetically inactivated in many human cancers and has a central role in the DNA damage response through the establishment of several complex regulatory loops involving p53, SIRT1 and E2F1 (Britschgi et al., 2006; Tseng et al., 2009). As shown by Naqvi et al., (2010) and Van Rechem et al., (2010) the p53-binding sequence lies in a region of the SIRT1 (SIRTUIN1) promoter that also binds the transcriptional repressor HIC1 tumor suppressor. Mohammad et al., (2011) have shown that the homozygous deletion and/or inactivation of HIC1 in mice results in major developmental defects and embryonic lethality. The same results were also reported by Dehennaut and Leprince, (2009). They showed that all copies of HIC1 are completely unmethylated and ubiquitously expressed in normal mammalian tissues (Dehennaut and Leprince, 2009). Fleuriel et al., (2009) proposed that epigenetical inactivation of HIC1 might “addict” cancer cells to altered survival and signaling pathways during the early stages of tumorigenesis by cooperating within a complex of HIC1-p53-SIRT1 regulatory loop. A HIC1-SIRT1-p53 circular loop in which hypermethylation of HIC1 represses the transcription of SIRT1 that deacetylates and inactivates p53 thus leading to HIC1 inactivation has been identified in cell and animal models. Hypermethylated HIC1 is an epigenetically regulated transcriptional repressor that functionally cooperates with p53 to suppress age-dependent development of cancer in mice (Chen et al., 2005). Tseng et al.(2009) proposed that deregulation of the HIC1-SIRT1-p53 loop in lung cancer patients is linked to poor prognosis. Yegnasubramanian et al., (2004) claimed that the aberrant DNA hypermethylation patterns may be the earliest somatic genome changes in prostate cancer for glutathione S-transferase pi (GSTP1), adenomatous polyposis coli (APC), RASSF1a, PTGS2, and multi-drug resistance gene 1 (MDR1) genes. The CpG islands for EDNRB, ESR1, CDKN2a, and hMLH1 genes exhibit low to moderate rates of hypermethylation and the CpG islands for DAPK1, TIMP3, MGMT, CDKN2b, p14/ARF, and CDH1 genes are unmethylated for prostate carcinomas (Yegnasubramanian et al., 2004). Our results showed moderate type hypermethylation for DAPK1 (33.3%) and hypomethlated profile for MGMT gene (8.3%) as reported by Yegnasubramanian et al., (2004).
In conclusion, we have investigated promoter hypermethylation status of six genes, p16, SFRP2, HIC1, DAPK1, and MGMT in 30 prostate carcinoma patients. Our results confirm the importance of methylation in the molecular pathogenesis of prostate adenocarcinomas, with the majority of tumors having a high rate of CpG islands methylated in HIC1, SFRP2, and DAPK1 gene promoters. Thus, tumoral tissue specific identification needs not only pathological diagnosis, but also oncogenetic and epigenetic analyses, for the correct diagnosis and case specific therapy in prostate carcinoma.
Footnotes
Disclosure Statement
There is no conflict on the current results.