
Abstract
X11α is a brain specific multi-modular protein that interacts with the Alzheimer's disease amyloid precursor protein (APP). Aggregation of amyloid-β peptide (Aβ), an APP cleavage product, is believed to be central to the pathogenesis of Alzheimer's disease. Recently, overexpression of X11α has been shown to reduce Aβ generation and to ameliorate memory deficit in a transgenic mouse model of Alzheimer's disease. Therefore, manipulating the expression level of X11α may provide a novel route for the treatment of Alzheimer's disease. Human X11α is encoded by the gene APBA1. As evidence suggests that X11α expression can be regulated at transcription level, we have determined the gene structure and cloned the promoter of APBA1. APBA1 spans over 244 kb on chromosome 9 and is composed of 13 exons and has multiple transcription start sites. A putative APBA1 promoter has been identified upstream of exon 1 and functional analysis revealed that this is highly active in neurons. By deletion analysis, the minimal promoter was found to be located between −224 and +14, a GC-rich region that contains a functional Sp3 binding site. In neurons, overexpression of Sp3 stimulates the APBA1 promoter while an Sp3 inhibitor suppresses the promoter activity. Moreover, inhibition of Sp3 reduces endogenous X11α expression and promotes the generation of Aβ. Our findings reveal that Sp3 play an essential role in APBA1 transcription.
Introduction
X11α, also known as Munc-18-interacting protein-1 (mint1), is a neuronal-specific adaptor protein that is implicated in a variety of neurobiological functions such as dendritic transport, copper homeostasis, and presynaptic functions. Since it is an adaptor protein, X11α mediates these functions by complexing via several protein-protein interacting domains. These include a Munc-18 interacting region, a CASK interacting region, a phosphotyrosine binding (PTB) domain, and two PSD-95/discs large/ZO-1 domains. Through its centrally located PTB domain, X11α interacts with the YENPTY motif of the cytoplasmic domain of APP. Studies from various laboratories have demonstrated that overexpression of X11α stabilizes APP and reduces Aβ levels in both cultured cells and transgenic mice APP [see reviews (Miller et al., 2006; Rogelj et al., 2006)]. Additionally X11α has been found to interact with a transcription factor fibrinogen silencer binding protein to suppress the human glycogen synthase kinase 3β (GSK3β) promoter (Lau et al., 2010). GSK3β is a candidate kinase for hyperphosphorylating tau in Alzheimer's disease. Therefore X11α may function as a protective agent against both Aβ production and tau hyperphosphorylation.
As overexpression of X11α reduces Aβ levels without causing any adverse phenotype in transgenic mice (Mitchell et al., 2010), enhancing expression of the neuronal specific X11α may be a potential therapeutic approach for Alzheimer's disease. At present, the mechanism(s) by which the expression of X11α is regulated remains elusive. However, there is evidence to suggest that X11α expression can be regulated at transcription level. First, X11α mRNA was only detected in brain but not in other tissues (Duclos et al., 1993; McLoughlin et al., 1999; Tomita et al., 1999). Second, developmental alteration of the Drosophila X11 homolog mRNA has been reported (Hase et al., 2002). Third, X11α mRNA has been found differentially expressed in various brain regions in mouse (Nakajima et al., 2001). As an initial step to unravel the transcriptional control of the human APBA1, the official gene symbol for the human X11α, we have cloned and characterized the human APBA1promoter.
Materials and Methods
Cell culture and transfection
HEK293 cells and Sprague Dawley rat cortical neurons were cultured as previously described (Lau et al., 2008). HEK293 cells and rat cortical neurons were transfected by using FuGENE 6 (Roche) and Lipofectamine 2000 (Invitrogen), respectively.
RNA ligase-mediated rapid amplification of cDNA ends
The transcription start sites of human APBA1 were determined by RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE). FirstChoice RACE-Ready Human Brain cDNA (Ambion) was used as a template for amplification with X11α RACE primer 4 (5′ TGCTGGCTGAGCGCGCCAGGCAT 3′) and 5’ RACE outer primer (5′ GCTGATGGCGATGAATGAACACTG 3′). For nested polymerase chain reaction (PCR), X11α RACE primer 5 (5′ GTCGGCCTCCACCGACTCGTTCA 3′) and 5′ RACE inner primer (5′ CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG 3′) were employed. PCR conditions consisted of initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s, with final extension 72°C for 7 min. The PCR products were cloned and sequenced.
Amplification of the human APBA1 promoter
The putative human APBA1 promoter was amplified from the human genomic clone RZPDB737B072039D (Imagenes) using X11α specific primer 1 (5′ CCTCAGCTTCCCGAGTAGCTAGGATTAC 3′) and X11α specific primer 2 (5′ ATCGGGTACCGTGCTGGGATTACAGGCGTGAG 3′). PCR conditions consisted of initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing and extension at 68°C for 2.5 min, with a final extension at 72°C for 7 min. The PCR products were cloned into pCR2.1 vector (Invitrogen) and sequence-verified.
Plasmid constructs
The putative human APBA1 promoter was subcloned into the promoterless firefly reporter vector pGL4.17[luc2/Neo] (Promega) to form p-1586Luc reporter construct. APBA1 promoter deletion constructs were generated either by PCR or by restriction digestion. Internal control pRL-CMV construct was purchased from Promega. Mammalian expression constructs of Sp1, Sp2, Sp3, and Sp4 were as described (Rotheneder et al., 1999).
Promoter reporter assay
Promoter luciferase reporter assays were performed as described (Yu et al., 2010). HEK293 cells and primary rat cortical neurons were transiently transfected with APBA1 promoter reporter and internal control pRL-CMV (Promega) constructs. Cells were harvested 48 h post-transfection for firefly/Renilla luciferase assay using a Dual-Glo Luciferase Assay System (Promega). The firefly luciferase activity was normalized to the corresponding Renilla luciferase activity.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed by using a gel shift assay system (Promega). Rat cortical neuron nuclear extract was isolated essentially as described (Yu et al., 2010). Double-stranded Sp3-A and Sp3-B probes were prepared by incubating Sp3-A-f 5′ GTCACGAGGGGCCAGCGAGCTA 3′+Sp3-A-r 5′ TAGCTCGCTGGCCCCTCGTGAC 3′ and Sp3-B-f 5′ AGGGGGTGGGGCCGCGAGACCGGA 3′+Sp3-B-r 5′ TCCGGTCTCGCGGCCCCACCCCCT 3′ in annealing buffer (20 mM Tris pH7.5, 10 mM MgCl2, 50 mM NaCl) at 80°C for 10 min and then cooled at room temperature. The probes were then labeled with [γ-32P]ATP by T4 polynucleotide kinase at 37°C for 1 h. The probes were added to 1X gel shift binding buffer either with or without nuclear extract. Supershift assay was performed by adding either anti-Sp1 or anti-Sp3 antibodies (Santa Cruz). The samples were resolved by a 4% non-denaturing polyacrylamide gel and visualized by autoradiography.
Semiquantitative reverse transcription (RT)-PCR, immunoblot analyzes, and Aβ enzyme-linked immunosorbent assays
Five days in vitro rat cortical neurons were treated either with vehicle (methanol) or with 100 nM mithramycin A for 48 h. Reverse transcription (RT)-PCR and immunoblot analyzes of the neurons were performed as described (Yu et al., 2010). PCR of X11α was performed by using the following two primers: 5′ CGCATGGACAGCTACGAGC 3′ and 5′ TAAGGCGAACGGATGGTC 3′. G3PDH was used as control. Primary antibodies for immunoblot analyzes were anti-X11α (Santa Cruz) and anti α-tubulin DM1A (Sigma). Secreted Aβ1-40 in the culture medium was measured by using an Aβ40 ELISA kit (Invitrogen).
Statistical analysis
Instat statistical software (GraphPad) was used for data analysis. Data are expressed as mean±standard deviation. Asterisk (*) indicates p<0.001.
Results
Determination of transcription start sites
To avoid premature termination that may occur in other classical transcription start site mapping techniques, RLM-RACE was used in this study. As shown in Figure 1A, no specific band was obtained in the primary PCR. However, a 0.4 kb DNA fragment was yielded in the nested PCR. This fragment was cloned and sequenced. All the clones sequenced contain the 5′ untranslated region of the human X11α cDNA. Interestingly, the inserts of the clones contain six different length DNA fragments, and the six termination points are closely located (Fig. 1B). This suggests that APBA1 contains multiple transcription start sites. In this study, the transcription start site nearest to the translation start codon of X11α is designated as +1.
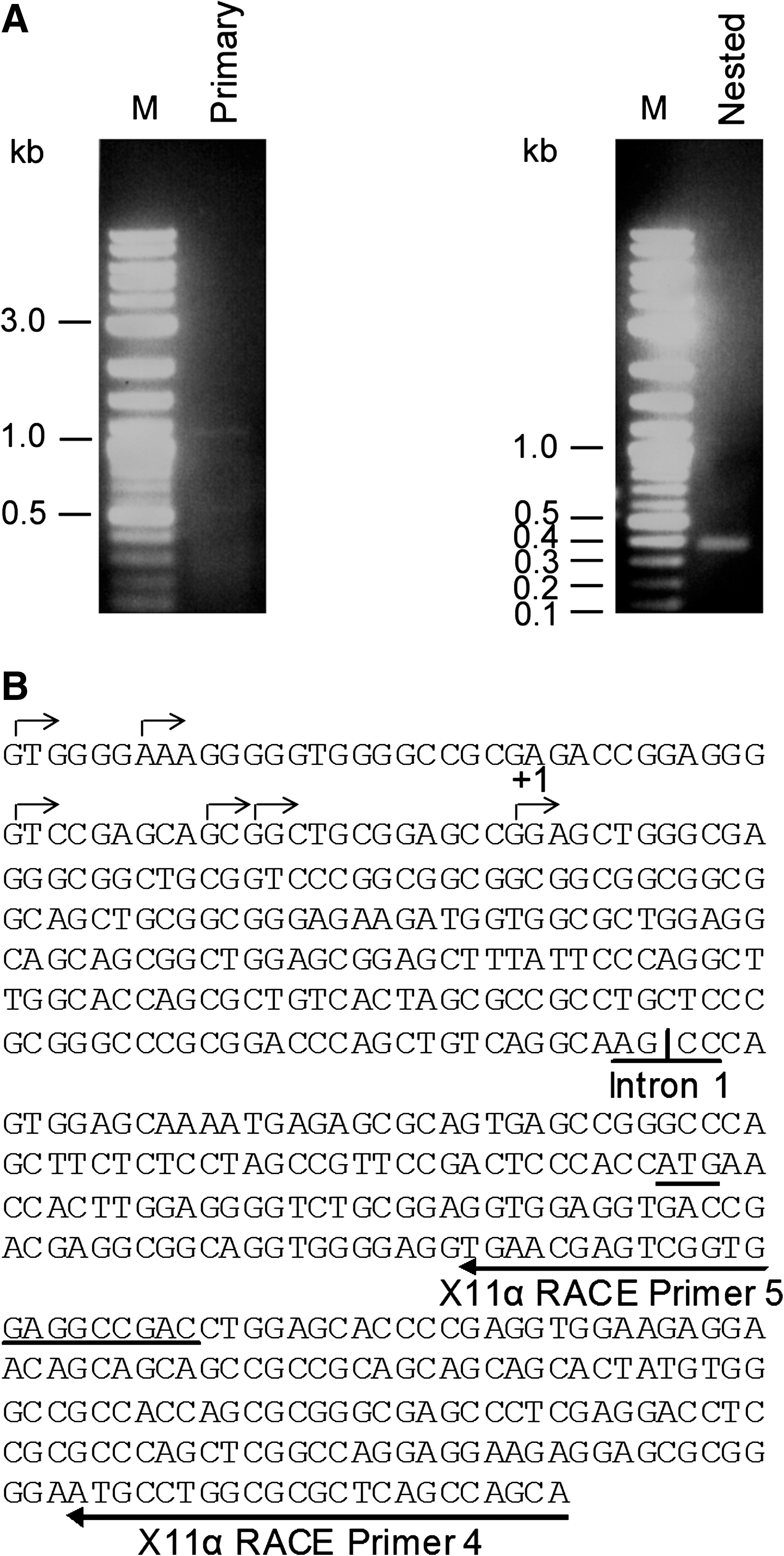
Mapping of the human APBA1 transcription start sites.
Genomic organization of the human APBA1
With the identification of the transcription start sites, we were able to locate exon 1 of the human APBA1. Together with information (accession no. NT_008470) from Genbank, the genomic structure of the human APBA1 has been deducted. APBA1 gene spans over 244kb on chromosome 9 (9q13-q21.1) and is organized into 13 exons and 12 introns (Fig. 2A, B). Classical GT/AG exon/intron junction organization is found in all the splice sites (Fig. 2C). The translation start and termination sites are located at exons 2 and 13, respectively.
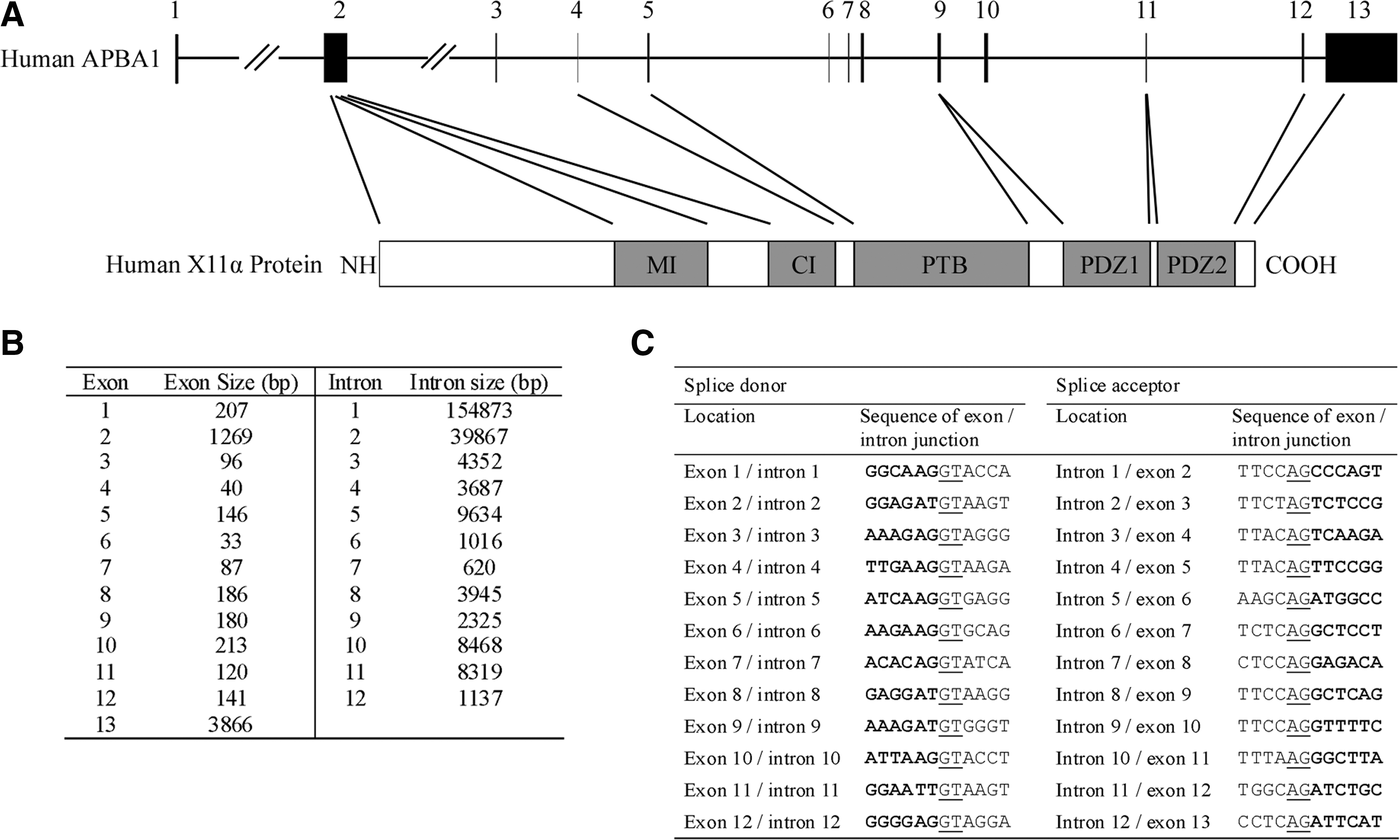
Genomic organization of the human APBA1.
Isolation and analysis of the putative human APBA1 promoter
In silico analysis by PROMOTER SCAN (Prestridge, 1995) revealed that a putative promoter region is located upstream of the APBA1 exon 1 (promoter score=63.77; cutoff=53). We therefore amplified a 1.6 kb fragment which contains part of the exon 1 and the putative APBA1 promoter, and cloned the DNA into the promoterless pGL4.17[luc2/Neo] vector (Fig. 3A). The APBA1 promoter reporter (p-1586Luc) and pRL-CMV normalization constructs were co-transfected into HEK293 cells and rat cortical neurons. Surprisingly, a significant fivefold increase in luciferase reporter activity was observed in HEK293 cells (n=12, p<0.001) (Fig. 3B). It has been reported that X11α mRNA is expressed in non-neuronal cells (Zhang et al., 2004). By RT-PCR, we confirmed that low level X11α mRNA is present in HEK293 cells (data not shown). In rat cortical neurons, the reporter activity of the p-1586Luc construct was 23-fold higher than the control (n=12, p<0.0001) (Fig. 3B). This finding indicates that the APBA1 promoter is more active in neurons than non-neuronal cells, and is in line with current knowledge regarding the expression of X11α. No reporter activity was observed when the promoter DNA was cloned in reverse orientation (i.e., p-1586Luc-R) in both HEK293 and neurons.

Sequence and functional analyses of the putative human APBA1 promoter.
Deletion analysis and EMSA of the human APBA1 promoter
To assess the functional region in the cloned APBA1 promoter, a series of 5′ deletion constructs were created. As shown in Figure 4A, all the deletion constructs from −1007 to −224 exhibited similar reporter activities to p-1586Luc. However, marked reduction in promoter activity was observed in p+14Luc. Therefore the sequence between −224 to +14 may contain the minimal promoter of APBA1 that is required for basal transcription activity. Sequence analysis by PROMO (
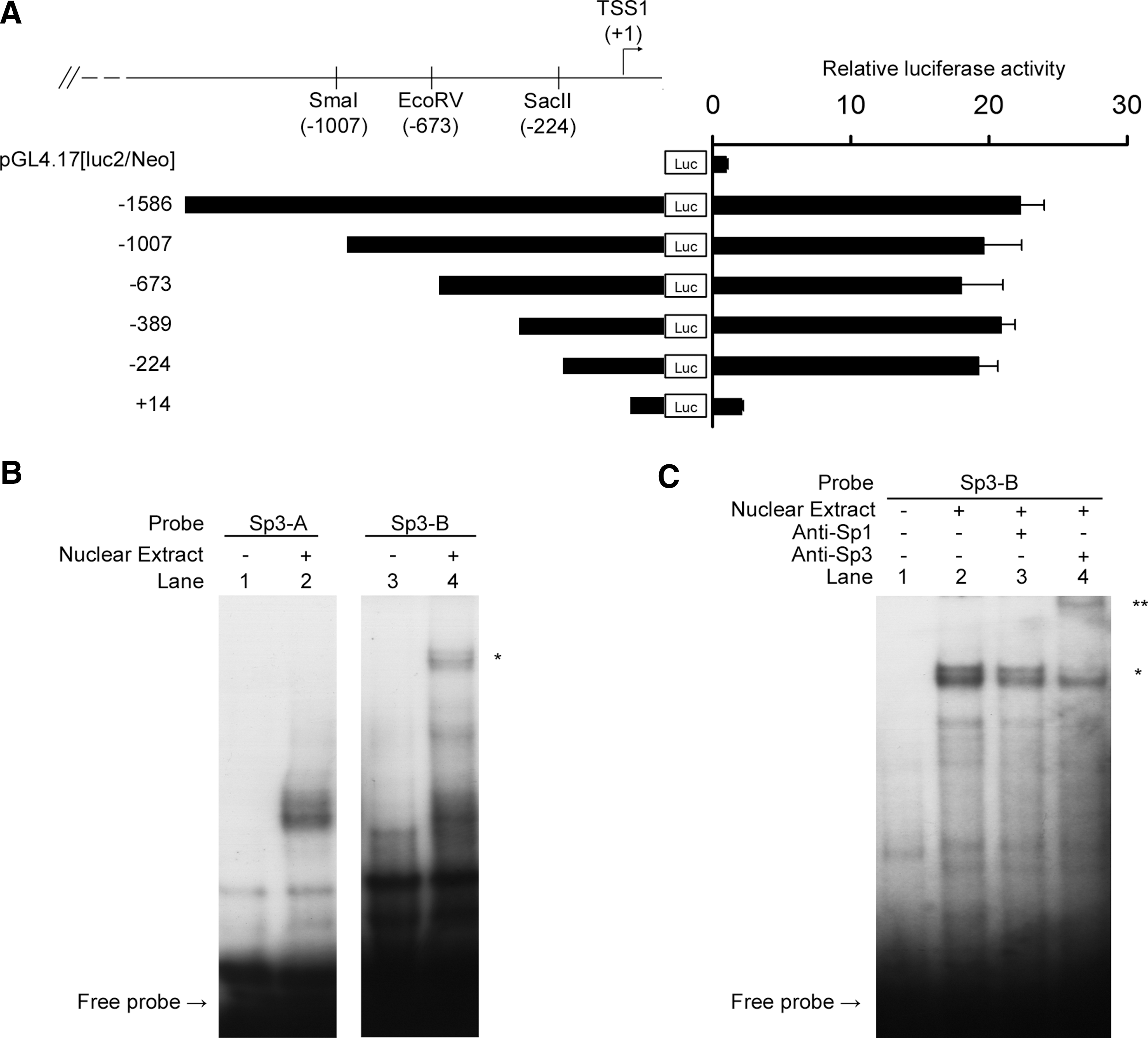
Deletion analysis and electrophoretic mobility shift assay (EMSA) of the human APBA1 promoter.
Sp3 influences APBA1 promoter activity
To examine if Sp3 alters the APBA1 promoter activity, p-1586Luc and various Sp family proteins were co-transfected to rat cortical neurons. Overexpression of Sp1 and Sp2 did not have any significant effect on the promoter. When co-transfected with Sp4, the promoter activity was slightly enhanced. However, a marked increase in promoter activity was observed in Sp3 co-transfected neurons (Fig. 5A). To further confirm the effect of Sp3, we treated p-1586Luc transfected neurons with mithramycin A which inhibits the binding of Sp3 to its target sequence (Yoo et al., 2002; Ryu et al., 2006; Xu et al., 2007). In the presence of mithramycin A, the APBA1 promoter activity was significantly suppressed (Fig. 5B).
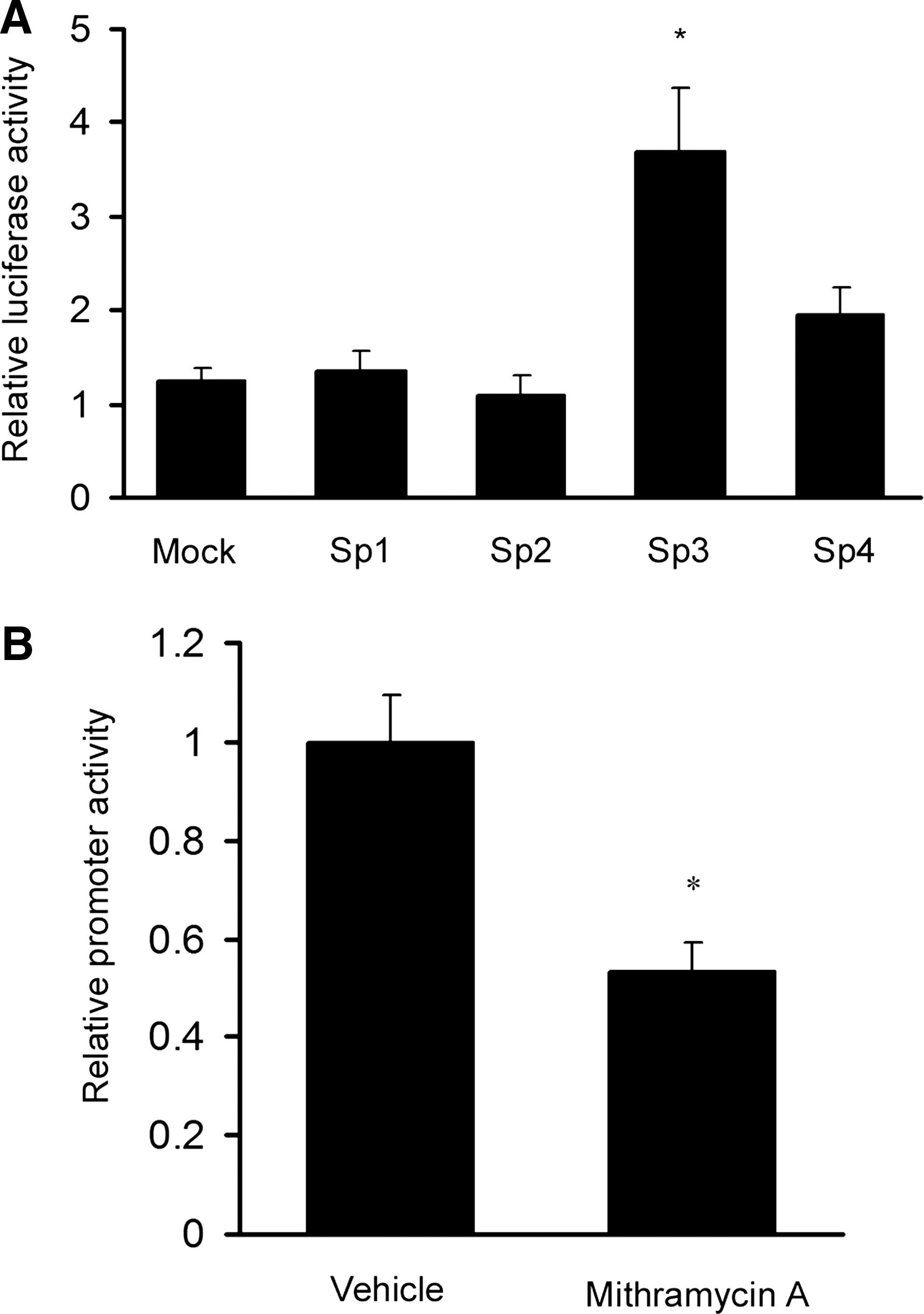
Effect of Sp3 on the APBA1 promoter transcriptional activity.
Inhibition of Sp3 suppresses the expression of X11α and enhances Aβ generation in neurons.
To investigate whether Sp3 regulates endogenous X11α expression, neurons were treated with the Sp3 inhibitor mithramycin A. The expression of both X11α mRNA and protein were reduced in the treated neurons (Fig. 6A, B). Such findings further confirm that Sp3 plays a role in regulating the expression of X11α. As X11α has been shown to reduce the generation of Aβ in various systems, we therefore measured Aβ level in the mithramycin A treated neurons. We found that the level of secreted Aβ1-40 significantly increased in mithramycin A treated neurons (Fig. 6C).
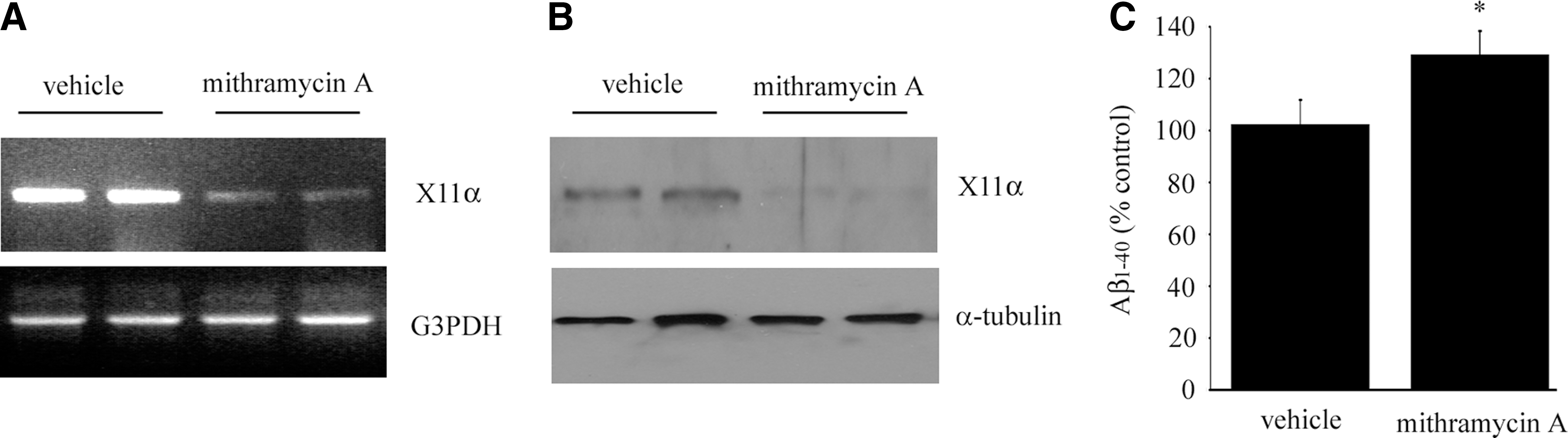
Inhibition of Sp3 by mithramycin A reduces the expression of X11α and enhances amyloid-β peptide (Aβ) generation in neurons. Rat cortical neurons were treated with either vehicle or 100 nM mithramycin A for 48 h. The levels of X11α mRNA and protein in the neurons were determined by
Discussion
To understand the mechanism by which X11α expression is controlled, we have cloned and characterized the promoter of APBA1. The promoter activity was examined in both neurons and non-neuronal cells. It has been reported that X11α mRNA and protein are mainly detected in neuronal tissues (Duclos et al., 1993; McLoughlin et al., 1999; Tomita et al., 1999). Our finding that the human APBA1 promoter is more active in neurons than in HEK293 cells is in line with the brain specific expression of X11α. This finding indicates that the promoter plays a role in controlling the neuronal expression of X11α. However, we cannot exclude the possibility that the expression X11α is regulated by other means. For example, several putative brain related miRNAs for X11α have been identified (Please see
In the present study, we have also determined the genomic structure of the human APBA1. Similar to many other eukaryotic genes, the first intron of X11α is much longer than the other introns in the gene (Kriventseva and Gelfand, 1999; Bradnam and Korf, 2008). Although the functional significance of long first introns is still not fully understood, many vertebrate orthologous first introns tend to be the most conserved intron (Chamary and Hurst, 2004; Vinogradov, 2006). It has been suggested that long first introns contain binding elements for regulatory proteins or RNAs. To avoid interference between bound factors, long introns are required for providing adequate spacing (Bradnam and Korf, 2008). Indeed, a number of studies have shown that early introns are essential for high expression of some genes (Mascarenhas et al., 1990; Palmiter et al., 1991; Jonsson et al., 1992; Rose and Last, 1997; Ho et al., 2001). It is therefore worthwhile to investigate the role of the first intron in the transcription of APBA1.
Deletion analysis has revealed that the GC-rich region between −224 to+14 contains the minimal APBA1 promoter. The core promoter architecture of the human APBA1 suggests that it is a null promoter as no recognizable TATA and initiator elements are found. It has been shown that the transcription of a null promoter may begin at multiple start sites [see review (Novina and Roy, 1996)]. Indeed, we have identified six closely located transcription start sites (+1 to −58) by RLM-RACE. This observation further suggests that the human APBA1 promoter belongs to the null category. Although there is no canonical TATA element, two Sp3 binding GC boxes were predicted within the minimal promoter. It has been suggested that GC box binding factors play important roles in the transcription of many neuronal TATA-less gene [see review (Butler and Kadonaga, 2002)], and Sp3 has been found to regulate GC-rich gene promoters (Hagen et al., 1992; Phillips et al., 2005; Abdelrahim et al., 2007; Chintharlapalli et al., 2007; Chadalapaka et al., 2008; Khalil et al., 2008; Valin et al., 2009). In fact, we have shown that Sp3 could stimulate the expression of the APBA1 promoter markedly but not Sp1, Sp2, and Sp4. We further confirmed that Sp3 binds to the GC-box located at −43 to −35 by EMSA. Postmortem analysis has revealed that Sp3 level is increased in the brains of Braak stages IV or VI of Alzheimer's disease patients (Boutillier et al., 2007). However, the exact role of Sp3 in Alzheimer's disease is largely unknown. It is noteworthy that X11α level is also found increased in Alzheimer's disease brains (Braak stages V or VI) and this has been suggested to play a beneficial role (Jacobs et al., 2006). Hence, it is possible that upregulation of Sp3 level enhances the expression of X11α which exerts protective effect against aberrant processing of APP. On the other hand, acetylation and sumoylation of Sp3 have also been shown to influence the transcription of some Sp3 target genes (Ammanamanchi et al., 2003; Spengler et al., 2005). Thus, investigation of the acetylation and sumoylation status of Sp3 in Alzheimer's disease and their effects on X11α expression may provide further insight into the roles of Sp3 in the disease.
Suppression of APP secretases has been proposed for lowering Aβ generation [see review (Dominguez et al., 2001)]. But APP is not their sole target and inhibition of the APP secretases has been found to interfere other cellular processes (Harrison et al., 2003; Searfoss et al., 2003; Ohno et al., 2004, 2006; Wong et al., 2004; Dominguez et al., 2005; Laird et al., 2005). It is therefore important to develop alternative approaches for reducing Aβ levels such as manipulating the expression of X11α. Hence, our study has set an essential basis for dissecting the transcriptional control of APBA1. In summary, we have determined the genomic structure of human APBA1, cloned and analyzed its promoter, and found that Sp3 is involved in regulating APBA1 transcription.
Footnotes
Acknowledgments
We thank Hans Rotheneder for mammalian expression constructs for Sp1, Sp2, Sp3, and Sp4. This work was supported by funds from the Research Grant Council Hong Kong and the CUHK direct grant scheme.
Disclosure Statement
The authors express no conflict of interest.