
Abstract
An understanding of cellular processes that determine the response to ionizing radiation (IR) exposure is essential to improve radiotherapy and to assess risks to human health after accidental radiation exposure. Exposure to IR induces a multitude of biological effects. Recent studies have indicated the involvement of epigenetic events in regulating the responses of irradiated cells. DNA methylation, where the cytosine bases in CpG dimers are converted to 5-methyl cytosine, is an epigenetic event that has been shown to regulate a variety of biological processes. We investigated the DNA methylation changes in irradiated TK6 and WTK1 human cells that differ in sensitivity to IR. The global DNA methylation alterations as measured by an enzyme-linked immunosorbent assay-based assay showed hypomethylation in both type of cells. Using an arbitrarily primed polymerase chain reaction (AP-PCR) approach, we observed time-dependent dynamic changes in the regional genomic DNA methylation patterns in both cell lines. The AP-PCR DNA methylation profiles were different between TK6 and WTK1 cells, indicating the involvement of differential genomic DNA responses to radiation treatment. The analysis of the components of the DNA methylation machinery showed the modulation of maintenance and de novo methyltransferases in irradiated cells. DNMT1 mRNA levels were increased in TK6 cells after irradiation but were repressed in WTK1 cells. DNMT3A and DNMT3B were induced in both cells after radiation treatment. TET1, involved in the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), was induced in both cells. This study demonstrates that irradiated cells acquire epigenetic changes in the DNA methylation patterns, and the associated cellular machinery are involved in the response to radiation exposure. This study also shows that DNA methylation patterns change at different genomic regions and are dependent on time after irradiation and the genetic background of the cell.
Introduction
Modifications to DNA comprise the epigenome of a cell. The epigenome changes through development and controls access to genes. Regulation of gene expression involves multi-layered mechanisms in which epigenetic modifications such as DNA methylation play a major role (Bird, 2002). DNA methyltransferase converts cytosine to 5-methyl cytosine in cytosine phosphodiester guanine (CpG) dimers, which are found in regions known as CpG islands (usually located in the promoter regions of genes). The proteins MeCP1 or MeCP2 bind to DNA with at least 12 methylated CpG dimers and block transcription (Bowen et al., 2004). Therefore, when CpG islands are hypermethylated, the gene is repressed/silenced, and similarly, when it is hypomethylated, transcription can take place and the gene is expressed. Such control is of significance for a wide range of biological processes, ranging from cellular differentiation during development, genomic imprinting, and X-chromosome inactivation to the maintenance of genome stability (da Rocha and Ferguson-Smith, 2004). Epigenetic regulation by CpG methylation has an important role in tumorigenesis. Disruption of epigenetic regulation during malignant transformation results in aberrant cellular proliferation (Baylin and Ohm, 2006). DNA methylation at promoter regions of genes involved in cell-cycle regulation and DNA repair leads to their abnormal epigenetic silencing in many cancers (Tessema et al., 2003).
Evidence is accumulating to suggest that the effects of radiation include epigenetic alterations, including altered DNA methylation. The potential role of epigenetic mechanisms in the regulation of the effects of IR has been reviewed (Ma et al., 2010). Methylation changes as determined by methyl-CpG immunoprecipitation were seen in irradiated MCF7 breast cancer cells (Kuhmann et al., 2011). Radiation induced changes in global DNA methylation patterns and repeat elements in AG01522 and RKO colon carcinoma cells have been reported (Goetz et al., 2011). The evaluation of irradiated GM10115 cells for global methylation showed the evidence of both hypomethylation and hypermethylation (Aypar et al., 2010). Irradiation leads to altered DNA methylation in the exposed mouse testis tissue (Tamminga and Kovalchuk, 2011). The effect of X-ray irradiation on methylation levels of p16, MGMT, and APC genes have been evaluated in colorectal cancers and normal colonic mucosa (Krakowczyk et al., 2010). The CpG methylation profiles of radiosensitive H460 and radioresistant H1299 human nonsmall cell lung cancer (NSCLC) cell lines revealed hypermethylated of many genes in radioresistant H1299 cells. In contrast, promoter CpG sites of many genes were hypomethylated in radiosensitive H460 cells (Kim et al., 2010). The impact of genomic methylation on radiation sensitivity of four colorectal carcinoma cell lines (HCT116, SW480, L174 T, and Co115) concluded that genomic hypomethylation results in enhanced radiation sensitivity (Hofstetter et al., 2010). Radiation and doxorubicin-resistant variant MCF-7DOX breast cancer cells harbor much lower levels of global DNA methylation than the MCF-7 cells (Luzhna and Kovalchuk, 2010). The maintenance of genomic DNA methylation under gamma-radiation stress is a dynamic process, and gamma-radiation treatment leads to DNA demethylation in mouse tissues (Batra et al., 2010).
The goal of this study was to correlate changes in the DNA methylation to radiation sensitivity by analyzing cells that are proficient or deficient in p53. The p53 protein is a key regulator of gene expression and is essential for maintaining genomic stability (Harvey et al., 1993). The p53 is implicated in DNA damage-induced G1 cell-cycle arrest (Kuerbitz et al., 1992), apoptosis (Lowe et al., 1993), and DNA repair (Bakalkin et al., 1994). We took advantage of two well-characterized lymphoblastoid cell lines TK6 and WTK1 that were derived from the same progenitor cell line WIL2, isolated from a single male donor. TK6 exhibits the wild-type p53 allele, while WTK1 is a p53 negative mutant (Amundson et al., 1993). WTK1 has been shown to acquire up to ten times more mutations at the thymidine kinase (tk) locus than TK6 after exposure to IR (Xia et al., 1994b). WTK1 cells are more efficient in recombinational repair and have higher resistance to X-irradiation-induced killing than TK6 cells (Xia et al., 1994a). Alterations in the expression of many nuclear genes have been suggested as contributing to the differences in the response of these cells to IR (Tsai et al., 2006). The changes in the DNA methylation of irradiated TK6 and WTK1 have not been reported earlier. This study was undertaken to investigate whether the radiation treatment has the ability to induce changes in the DNA methylation levels of these cells and to investigate the status of DNA methylation machinery in these cells after radiation treatment.
Materials and Methods
Cell culture and maintenance
The human lymphoblast cell lines TK6 and WTK1 were obtained from Dr. Howard Liber, Colorado State University, Fort Collins, CO. These cells were exponentially grown in suspension in a T75 flask using RPMI 1640 supplemented with 10% fetal bovine serum (Invitrogen, Grand Island, NY), 100 mg/mL streptomycin, and 100 U/mL penicillin. The cell cultures were maintained at a density of 2–5×105 cells/mL in a 37°C incubator with 5% CO2.
Ionizing radiation treatment
Irradiation of 3×106 cells was performed with a RAD Source 2000 X-ray Biological irradiator (Alpharetta, GA). The cells were seeded at a density of 3×106 per mL, and a dose of 2 Gy at a dose rate of 1.7 Gy/min was administered at room temperature.
DNA isolation and modification
Genomic DNA was isolated from 5×106 exponentially growing cells with DNeasy kit (Qiagen) according to the protocol provided by the supplier. The genomic DNA was modified with sodium bisulfite treatment according to a published protocol (Herman et al., 1996). Briefly, 5 μg of genomic DNA diluted in 50 μL of water was used as the starting material. Two microgram genomic DNA was denatured in 2 N NaOH at 37°C for 15 min. and incubated for 16 h at 50°C in 3 M sodium bisulfite and 10 mM hydroquinone (Sigma-Aldrich, St Louis, MO) pH 5.5. The modified DNA was purified with a Wizard DNA purification kit (Promega, Madison, WI) according to the manufacturer's recommendations. The modified DNA was treated with 3 N NaOH for 5 min at room temperature followed by precipitation. The modified DNA was quantified after purification to determine the DNA loss during modification or purification, and to calculate a defined quantity to be used in the polymerase chain reaction (PCR). Alternatively, ∼200 ng of DNA was modified with an EpiTect Bisulfite Kit (Qiagen, Valencia, CA).
Arbitrarily primed polymerase chain reaction
The arbitrarily primed polymerase chain reaction (AP-PCR) method was performed as described by Gonzalgo et al., (1997). Briefly, 100 ng of sodium bisulfite-treated genomic DNA was amplified in 50 μL of total volume, with 50 pmol of a single primer (5′ AACCCTCACCCTAACCCCGG 3′) as sense and antisense, 200 μM each of four deoxynucleotide triphosphates (dNTPs), 1.6 U Jump Start™ Taq polymerase (Sigma), 4 mM MgCl2, and 10 mM Tris–HCl buffer (pH 8.0). The AP-PCR program utilized 5 low-stringency cycles (94°C for 30 s, 40°C for 60 s, 72°C for 90 s) followed by 27 increased-stringency cycles (94°C for 15 s, 55°C for 15 s, 72°C for 60 s). Low-stringency PCR permits the identification of differentially methylated bands. The random association of primers with genomic DNA at low annealing temperatures produced multiple PCR fragments detected by 2% agarose gel electrophoresis after staining with ethidium bromide. For methylation-specific amplification, 1 U of Tsp509I restriction endonuclease (New England BioLabs, Ipswich, MA) was added to the reaction. All the amplification reactions were performed in an MJ Research PTC 200 thermocycler. The AP-PCR experiments were repeated three times, and there was negligible variability among the experiments.
Global DNA methylation quantification assay
Global DNA methylation levels were quantified with a commercially available DNA methylation quantification kit (Epigentek Inc., Farmingdale, NY). The use of the Epigentek kit to quantify global DNA methylation has been successfully demonstrated (Batra et al., 2010). In this assay, methylated DNA is immobilized on a strip and is recognized by 5-methylcytosine antibody followed by quantification through enzyme-linked immunosorbent assay (ELISA). Results were expressed as an absolute percentage of 5-methylcytosine. Briefly, 28 μL of DNA binding solution and 2 μL (200 ng) of sample DNA were incubated at 37°C for 2 h followed by a second incubation (to evaporate the solution) at 60°C for 20–30 min. For blank, 30 μL of DNA binding solution was used instead of DNA. To each dried well, 150 μL of blocking solution was added followed by incubation at 37°C for 30–45 min. 150 μL of wash buffer and capture antibody (1 μg/mL) was then added, and samples were incubated at room temperature for 60 min. Wells were washed with wash buffer, and detection antibody was added. The samples were incubated at room temperature for 30 min. Wells were washed 5 times with 150 μL of wash buffer. 100 μL of the developing solution was added to each well, and the plate was incubated at room temperature for 2–10 min in the dark. Color development was monitored in the sample and the control well. Finally, the reaction was stopped using stop solution (50 μL), and the absorbance was read on a microplate reader at 450 nm (Multiskan MCC/340; Thermo Scientific, West Palm Beach, FL). Results were calculated as percent of methylated DNA.
RNA isolation
The control (mock irradiated) and irradiated cells were counted with a hemocytometer. Approximately 5×106 cells were pelleted by centrifugation at 1500 rpm for 5 min, and washed with 1 mL Dulbecco's phosphate-buffered saline without MgCl2 and CaCl2 (Invitrogen, Carlsbad, CA). Total RNA was isolated with RNeasy™ kit (Qiagen, Valencia, CA). The quantity and quality of the total RNA was measured on the NanoDrop 2000 (Thermo Scientific, West Palm Beach, FL) and by analysis on 2% agarose gels stained with ethidium bromide on BioSpectrum® Imaging System (UVP, Upland, CA).
Gene targets
Assays on demand for DNA (cytosine-5)-methyltransferase 1 (DNMT1), DNA (cytosine-5)-methyltransferase 3A (DNMT3A), DNA (cytosine-5)-methyltransferase 3 beta (DNMT3B), and ten-eleven translocation 1 (hTPM1) genes were purchased from Applied Biosystems. RNA samples for gene expression analysis were normalized based on the TaqMan Gene Expression Assays for human endogenous hypoxanthine phosphoribosyltransferase (HPRT) gene.
Reverse transcription and cDNA synthesis reactions
Total RNA was treated with DNAse before reverse transcription in order to avoid genomic DNA contamination. The cDNA were generated from total RNA with random hexamer primers using a cDNA synthesis Kit from Applied Biosystems, following the recommendations of the manufacturer. The reaction contained RNA samples, 50 nM random hexamers, 1X reverse transcriptase (RT) buffer, 0.25 mM each of dNTPs, 3.33 U/μL MultiScribe™ reverse transcriptase, and 0.25 U/μL RNase inhibitor. The 15 μL reactions were incubated in Techne TC-312 thermocycler (Burlington, NJ) for 30 min at 16°C, 30 min at 42°C, 5 min at 85°C and then cooled at 4°C. All reverse transcriptase reactions, including no-template controls and RT minus controls, were run in duplicate.
Quantitative real-time polymerase chain reaction and data analysis
Quantitative real-time polymerase chain reaction (QPCR) was performed on an Applied Biosystems 7900HT Sequence Detection System by using a standard TaqMan PCR kit protocol as previously described (Chaudhry, 2009). Briefly, the 10 μL PCR contained 0.67 μL RT product, 1X TaqMan Universal PCR Master Mix, 0.2 μM TaqMan probe, 1.5 μM forward primer, and 0.7 μM reverse primer. The reactions were incubated in a plate at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The relative expression values of cycle thresholds were calculated by using the comparative delta cycle threshold, ΔΔCT method (Livak and Schmittgen, 2001) by normalization to the control HPRT and to the control nonirradiated sample. Control sample was used as the calibrator to calculate the relative expression and log2 values. The threshold cycle (CT) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold. Statistical significance was determined using analysis of variance. The statistics and data analysis was performed with ABI prism and GraphPad Prism 5 software, both licensed to the University of Vermont. CT values of all target genes in each cell lines were statistically evaluated using a one-way t-Test (p<0.05). The experiments were repeated three times, and the differences in all the miRNA were statistically significant between the cells treated with radiation.
Results
Analysis of global DNA methylation in irradiated cells
We determined the levels of global DNA methylation in 2 Gy irradiated TK6 and WTK1 cells in a time-course experiment using an ELISA based commercially available kit. WTK1 cells had much higher methylated DNA content as compared with TK6 cells (Fig. 1). After irradiation, changes in the global methylation were observed in both cell lines as early as 2 h postradiation treatment. DNA methylation levels continued to decline in WTK1 cells until 8 h, followed by an increase in the percentage of methylated DNA until the 12 h time point postirradiation. The percentage of methylated DNA in irradiated WTK1 cells returned to the background levels at the 24 h time point. The DNA methylation levels were initially decreased in TK6 cells after irradiation followed by a steady increase until 24 h time period. The difference in the DNA methylation between the irradiated TK6 and WTK1 cells was statistically significant (p=0.03).

Assessment of global DNA methylation levels in TK6 and WTK1 cells. The cells were irradiated with 2 Gy dose of X-rays and the genomic DNA was isolated at 0, 2, 4, 8, 12, and 24 h. DNA methylation was measured with an ELISA kit employing 5-methylcytosine antibody. The results were expressed as a percentage of methylation. The error bars indicate the standard error of the mean (SEM) for three independent experiments. ELISA, enzyme-linked immunosorbent assay.
Analysis of regional DNA methylation with AP-PCR in irradiated TK6 and WTK1 cells
To determine the genome-wide methylation fingerprint of irradiated cells in a time-course experiment, we exploited the AP-PCR technique applied to bisulfite-treated DNA template prepared from these cells. We introduced a modification in the AP-PCR protocol by adding heat-stable restriction enzyme Tsp509I digestion during the AP-PCR. The Tsp509I digest the unmethylated DNA leaving behind the methylated DNA as a template for AP-PCR amplification. Tsp509I cuts at AATT sites and has been used to analyze the efficiency of sodium-bisulfite-mediated DNA conversion (Goel et al., 2003). The cleavage of unmethylated template is based on the presence of CpG dinucleotides in the sequence context AATCG or AACCG. After sodium bisulfite conversion, these sequences will be converted to either AATCG (CpG methylated) or AATTG (CpG unmethylated). Only unmethylated CpG sites result in a recognition site after sodium bisulfite treatment, allowing cleavage of the template. We used Tsp509I directly in the PCR because of its optimal incubation temperature of 65°C, heat stability, and high activity under different buffering conditions. During each PCR cycle, the enzymatic activity of Tsp509I results in the cleavage of PCR products that originate from unmethylated sodium bisulfite-converted DNA, whereas methylated sequences are not cut by the enzyme.
We first examined the genomic DNA methylation profile in TK6 cells irradiated with 2 Gy of X-rays. The irradiated cells were sampled at 0, 2, 4, 8, 12, and 24 h after treatment, and the isolated DNA was subjected to AP-PCR in either the absence or presence of Tsp509I restriction endonuclease (Fig. 2). Only selected populations of DNA fragments enriched with sequences containing either methylated or unmethylated sites are amplified with this approach. DNA fingerprints of samples with gain/loss bands or decreased/increased band intensity were observed. There was negligible variability among repeated AP-PCR experiments. The DNA amplification fingerprint was very different in the time-course samples amplified with AP-PCR in the absence of Tsp509I as compared with the samples that were amplified by including Tsp509I in the reaction. The inclusion of Tsp509I in the PCR permitted the detection of methylated genomic regions that were otherwise undetectable because of their overall low abundance in the genome. There were dynamic changes in the AP-PCR fragment patterns as the cells recovered from the damage induced by radiation treatment. Alterations in the DNA methylation of specific genomic regions were evident as early as 2 h after radiation exposure. Interestingly the data indicated differential genomic DNA methylation as the cells progressed through time.
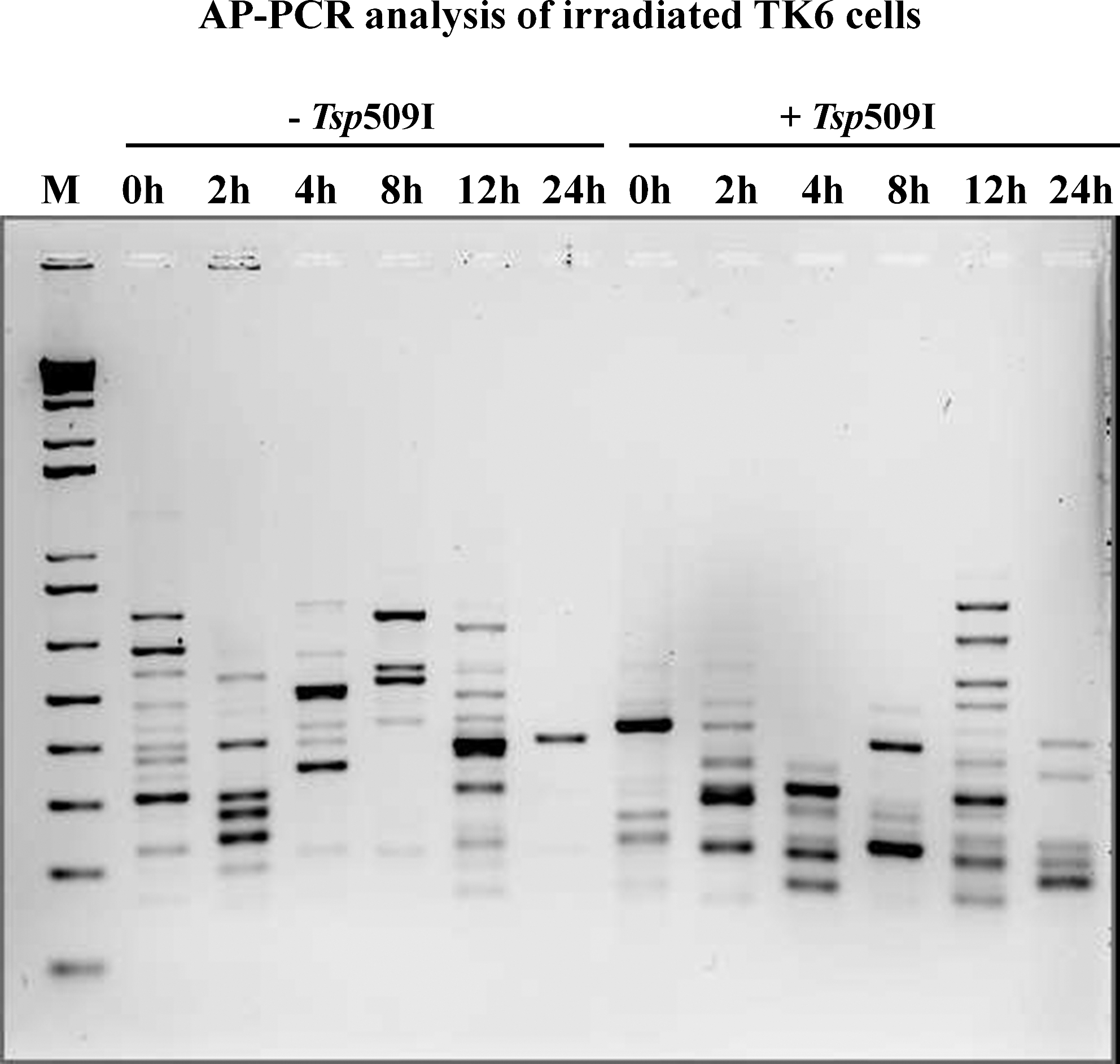
AP-PCR analysis of sodium bisulfite-treated DNA isolated from irradiated TK6 cells at various time points. The “-Tsp509I” indicates samples from the amplification reactions without the additions of heat resistant restriction enzyme Tsp509I during the PCR. The “+Tsp509I” indicates samples from the amplification reactions with the additions of Tsp509I during the PCR. The PCR products were electrophoresed in a 2% agarose gel and stained with ethidium bromide. The lane marked “M” is 1 Kb ladder, molecular weight standard from Invitrogen Inc. AP-PCR, arbitrarily primed polymerase chain reaction.
We performed the same experiment with the irradiated WTK1 cells. The AP-PCR fingerprint shown in Figure 3 identified that the amplified DNA patterns were different among the time-course samples analyzed with or without Tsp509I enzyme. The presence of Tsp509I allowed the detection of methylated regions in the WTK1 genome. As observed with TK6 cells, the WTK1 also exhibited dynamic alterations in the genomic DNA methylation profiles. The DNA methylation changes were seen throughout the time-course experiment, suggesting that the genomic methylation status changes as the cells recover from the radiation induced damage. Both hypermethylation and hypomethylation were observed in irradiated WTK1 cells over time.
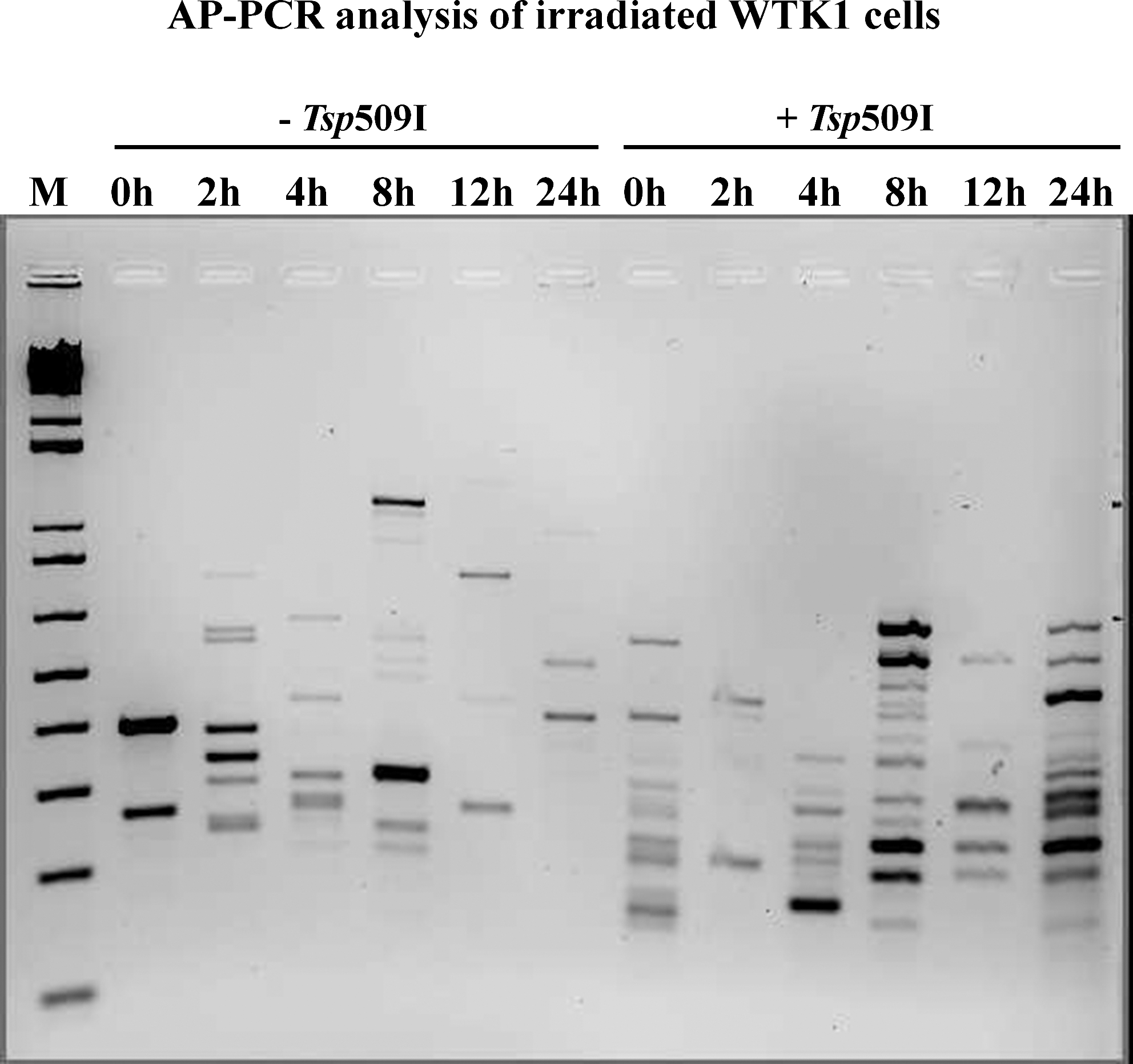
AP-PCR analysis of sodium-bisulfite-treated DNA isolated from irradiated WTK1 cells at various time points. The “-Tsp509I” indicates samples from the amplification reactions without the additions of heat-resistant restriction enzyme Tsp509I during the PCR. The “+Tsp509I” indicates samples from the amplification reactions with the additions of Tsp509I during the PCR. The PCR products were electrophoresed in a 2% agarose gel and stained with ethidium bromide. The lane marked “M” is a 1 Kb ladder, molecular-weight standard from Invitrogen Inc.
Finally, we compared the DNA methylation fingerprint in irradiated TK6 and WTK1 cells (Fig. 4). We found that there were differences in the AP-PCR amplified DNA profiles among the two cell lines at all the time points examined, indicating the involvement of methylation changes in different regions of the genome of these cells. Although AP-PCR is not quantitative, there was evidence of both hypermethylation and hypomethylation as observed by the intensity of various amplified DNA fragments from both TK6 and WTK1 irradiated cells during the 24 h time period.

AP-PCR analysis of sodium-bisulfite-treated DNA isolated from irradiated TK6 and WTK1 cells at various time points. The amplification reactions were carried out in the presence of heat-resistant restriction enzyme Tsp509I during the PCR. The PCR products were electrophoresed in a 2% agarose gel and stained with ethidium bromide. The lane marked “M” is a 1 Kb ladder, molecular-weight standard from Invitrogen Inc.
Modulation of DNA methylation genes in TK6 and WTK1 cells
We first investigated the changes in the expression levels of maintenance DNA methyltransferase DNMT1 in TK6 and WTK1 cells exposed to 2 Gy dose of X-rays. The irradiated cells were harvested at 0, 2, 4, 8, 12, and 24h, and the DNMT1 expression analysis was done by QPCR. DNMT1 mRNA levels were increased in TK6 cells after irradiation and reached the maximum values at 8 h time point (Fig. 5A). The DNMT1 expression levels started to decline after 8 h postirradiation and then returned to the baseline levels by the 24 h time period. In contrast, DNMT1 was repressed in WTK1 after radiation treatment (Fig. 5A). The difference in the expression of DNMT1 between the irradiated TK6 and WTK1 cells was statistically significant (p=0.02).
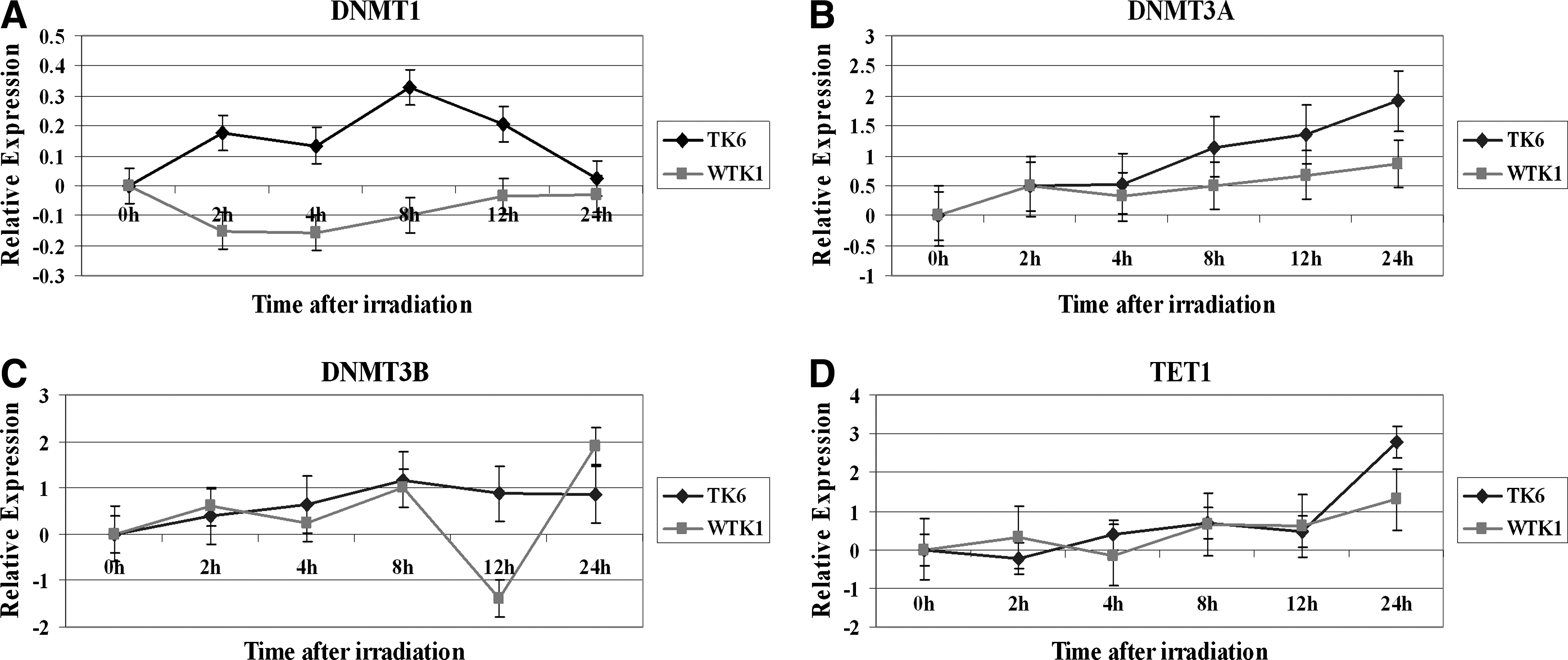
Modulation of DNMT1, DNMT3a, DNMT3b, and TET1 mRNA in irradiated TK6 and bystander cells. The cells were treated with 2 Gy X-rays dose, and the total RNA was isolated at various times, converted to cDNA, and then subjected to real-time PCR analysis using commercially available assays on demand for these genes. The relative expression, shown as Log2 values, was computed at 0, 2, 4, 8, 12, and 24 h time points.
The assessment of the expression of de novo DNA methyltransferase DNMT3A in X-rays-treated TK6 cells indicated an upregulation in its levels. The induction of DNMT3A continued throughout the 24 h time period after irradiation of TK6 cells and reached its maximum at the 24 h time point (Fig. 5B). Although DNMT3A was also induced in WTK1 cells after irradiation, its induction was much less as compared with the TK6 cells (Fig. 5B). The difference in the expression of DNMT3A between the irradiated TK6 and WTK1 cells was not statistically significant (p=0.05).
We next examined the modulation of the other de novo DNA methyltransferase DNMT3B in irradiated TK6 and WTK1 cells. There was a steady increase in the expression of DNMT3B in both cell lines after exposure to IR (Fig. 5C). A sharp reduction in the DNMT3B levels was observed at the 12 h time point in irradiated WTK1 cells followed by an induction at the 24 h time point. The difference in the expression of DNMT3B between the irradiated TK6 and WTK1 cells was not statistically significant (p=0.58).
Modulation of TET1 in irradiated TK6 and WTK1 cells
We finally examine the status of the expression of TET1 gene in irradiated TK6 and WTK1 cells. TET1 converts 5-methylcytosine (5-mC) in genomic DNA to 5-hydroxymethylcytosine (5-hmC). The pattern of TET1 expression was similar in TK6 and WTK1 cells. A noticeable difference in the induction of TET1 was seen at the 24 h time point where the level of the induction of TET1 was much higher in irradiated TK6 cells as compared with WTK1 cells (Fig. 5D). The difference in the expression of TET1 between the irradiated TK6 and WTK1 cells was not statistically significant (p=0.46).
Discussion
IR is a therapeutic agent for many cancers and is an important diagnostic tool. On the other hand, IR is a genotoxic agent that can affect a variety of cellular processes. Accumulation of DNA damage caused by IR in conjunction with disrupted cellular regulation processes can lead to many biological effects. It is suggested that epigenetic factors including DNA methylation could be involved in these processes. We determined the effect of IR treatment on the methylation of genomic DNA of two related cell lines that differ in p53 status. We examined the global methylation profile of DNA isolated from TK6 (p53 positive) and WTK1 (p53 negative) human lymphoblasts cells exposed to X-rays. These two cell lines are derived from the same individual, differ in their ability to rejoin DNA double-strand breaks (the most lethal damage to the cell) and sensitivity to radiation (Evans et al., 2001). WTK1 cells are more resistant to the cytotoxic effects of radiation than TK6 cells (Evans et al., 2001), thus providing a suitable model to examine the role of epigenetic factors in determining radiation sensitivity.
Radiation-induced alterations in global and regional genomic DNA methylation
We first assessed the global DNA methylation status in irradiated TK6 and WTK1 cells and discovered time-dependent differences in DNA methylation levels between the two cell lines. The global DNA methylation alterations as measured by an ELISA-based assay showed hypomethylation in WTK1 cells and hypermethylation in TK6 cells. The analysis of irradiated GM10115 cells for global methylation showed the evidence of both hypomethylation and hypermethylation (Aypar et al., 2010). Hypermethylation of many genes in the radioresistant H1299 human NSCLC cell line was observed, and many genes were hypomethylated in radiosensitive H460 cells (Kim et al., 2010). Genomic hypomethylation results in enhanced radiation sensitivity of colorectal carcinoma cell lines (Hofstetter et al., 2010). Radiation resistant variant MCF-7 breast cancer cells have been shown to harbor much lower levels of global DNA methylation than the MCF-7 cells (Luzhna and Kovalchuk, 2010). Other studies have reported that gamma-radiation treatment leads to DNA demethylation in mouse tissues (Batra et al., 2010). The radiation treatment results in loss of genomic DNA methylation in vertebrate animals (Koturbash et al., 2006a, Koturbash et al., 2006b, Loree et al., 2006).
We further scanned the genome of irradiated TK6 and WTK1 cells with AP-PCR technique to assess changes in the regional genomic methylation. The detection of locus specific methylation is accomplished by distinguishing the methylated DNA from unmethylated DNA with sodium-bisulfite-mediated conversion (Frommer et al., 1992). Methylation of cytosine in DNA protects against the conversion of cytosine to uracil during sodium bisulfite treatment. Unmethylated cytosine is converted to uracil by bisulfite treatment, and uracil is substituted by thymidine during PCR-mediated DNA amplification (Chaudhry, 2010). However, methylcytosine appears as cytosine after PCR. Bisulfite treatment of DNA is followed by several techniques, such as methylation-specific PCR (Herman et al., 1996), denaturing high-performance liquid chromatography (Omaruddin and Chaudhry, 2010), and many other approaches (Chaudhry, 2010) for DNA methylation analysis.
In general, DNA restriction is a common technique for methylation analysis. Methylation-sensitive restriction endonucleases have previously been used to digest nonbisulfite treated unmethylated template before PCR (Oakes et al., 2006, Oakes et al., 2009). The major disadvantage of this pre-PCR restriction is that even minor remnants of nondigested DNA will be amplified, leading to false-positive results. To overcome these limitations, we employed Tsp509I digestion of unmethylated background DNA during PCR cycling leading to specific detection of methylated DNA. Tsp509I heat stability allows for the digestion of undesired amplicons as they appear in each PCR cycle. The result is a strong bias for the amplification of bisulfite-converted methylated DNA. We combined the Tsp509I strategy with the well-known AP-PCR technique. The enrichment of methylated DNA provided a clearer picture of the extent of DNA methylation in irradiated cells. Our data show that the maintenance of genomic DNA methylation under radiation stress is a dynamic process. We found that different genomic regions were methylated at different times in irradiated TK6 and WTK1 cells, suggesting the regulation of cellular processes as cells survive the DNA damage.
Modulation of DNA methylation machinery in irradiated cells
DNA methylation at the C5 position of CpG dinucleotides is carried out by either maintenance methyltransferase or de novo methyltransferases. Maintenance methylation activity is necessary to preserve DNA methylation after every cellular DNA replication cycle. DNA nucleotide methyltransferase 1(DNMT1), is the maintenance methyltransferase that is responsible for copying DNA methylation patterns to the daughter strands during DNA replication (Jurkowska et al., 2011). DNMT3a and DNMT3b are the de novo methyltransferases that set up DNA methylation patterns early in development (Jurkowska et al., 2011). We examined the role of DNA methylation machinery in 2 Gy irradiated TK6 and WTK1 cells (Fig 5). The expression levels of DNMT1 were upregulated in TK6 cells but were downregualted in WTK1 cells. De novo DNA methyltransferases DNMT3a and DNMT3b were upregualed in both cells. An increased expression of DNMT1 and DNMT3b in 2 Gy irradiated pancreatic cancer cells was reported, but a decreased DNMT3b mRNA expression was observed after 4 Gy irradiation (Ma et al., 2011). A reduction in the levels of DNMT1, DNMT3a, and DNMT3b was seen in rat mammary tissue after X-ray irradiation (Loree et al., 2006). Other studies have shown that the loss of DNA methylation was paralleled by a significant decrease in the levels of DNMT1, DNMT3a, and DNMT3b in animals (Koturbash et al., 2006a). A significant reduction in the levels of DNMT1 and DNMT3a DNA methyltransferases and a concurrent increase in the levels of the DNMT1 in bystander tissues were observed. (Koturbash et al., 2006b). Most of these studies examining the role of DNA methylation machinery have been done in vertebrate animals. Our study on human cells differing in p53 has identified the modulation of DNA methytransferases after treatment with IR. p53 has been linked to the control of epigenetic regulation by genomic methylation. p53 negatively regulates DNMT1 expression by forming a complex with specificity protein 1 (Sp1) protein and chromatin modifiers on the DNMT1 promoter (Lin et al., 2010). p53 has also been shown to suppresses the histone methyltransferase EZH2 gene (Tang et al., 2004). Deletion of p53 results in an increase of DNMT1 mRNA suggesting relief of p53-mediated DNMT1 repression (Peterson et al., 2003). p53 controls DNMT1 transcription through direct DNA binding. Treatment with IR diminishes this binding, concomitant with increase in DNMT1 expression levels (Peterson et al., 2003). Our data suggest that IR-mediated activation of p53 in TK6 cells possibly relieves transcriptional repression of the DNMT1 gene, leading to its higher expression levels. Since the de novo DNA methyltransferases DNMT3a and DNMT3b are not under p53 control, their expression is not different in p53 positive TK6 and p53 negative WTK1 cells after radiation treatment.
Modulation of TET1 in irradiated TK6 and WTK1 cells
The cellular role of the modified form of cytosine 5-hydroxymethylcytosine (5-hmC) is unknown. It is suggested that the function of 5-hmC in epigenetics may be different from 5-methylcytosine (5-mC) (Kriaucionis and Heintz, 2009). TET1 is a 2-oxoglutarate (2OG)- and Fe(II)-dependent enzyme that converts 5-mC to 5-hmC (Tahiliani et al., 2009). TET1 potentially plays a role in epigenetic regulation. The role of TET1 in radiation-induced cellular response has not been determined. It is unknown whether the radiation treatment can change 5-methylcytosine to 5-hydroxymethylcytosine and, thus, epigenetically regulate cellular functions. We examined the expression of TET1 in irradiated TK6 and WTK1 cells harvested at various times by real-time reverse transcriptase–PCR. TET1 was upregulated in both types of cells at late time points after irradiation. An increased level of TET1 could increase cellular 5-hmC levels and could lead to hypomethylation.
Summary
The data presented here show that the radiosensitive TK6 and radioresistant WTK1 cells exhibit changes in the genomic DNA methylation patterns after exposure to IR. Furthermore, these changes are time dependent and affect different regions of the genome at different times in these cells. There were variations in DNA methylation patterns between cell lines differing in p53 gene status. The maintenance and de novo DNA methyltransferases are modulated in these cells after irradiation. The expression levels of TET1 are also modulated in these cells. These observations indicate that DNA methylation machinery is disturbed in TK6 and WTK1 cells after irradiation. DNA methylation could be a possible mechanism differentiating radiation responses between p53 negative and wild-type cell lines. The response of cells to radiation is very complex and involves the participation of many genes. We previously identified large-scale gene expression alterations in human cells exposed to IR (Chaudhry, 2006b). Later studies from our laboratory identified the contribution of microRNA (miRNA) in cellular responses to IR treatment (Chaudhry, 2009, Chaudhry et al., 2010a, Chaudhry et al., 2010b). In the current investigation of epigenetic factors DNA methylation has been identified, which might be responsible for controlling cellular responses after radiation exposure.
Footnotes
Acknowledgments
The authors thank Dr. Howard Liber, Colorado State University, Fort Collins, CO, for providing TK6 and WTK1 cells. The authors are thankful to the DNA analysis facility, University of Vermont, for assistance with the real-time PCR experiments. This work was supported by a research incentive grant from the College of Nursing and Health Sciences, University of Vermont.
Disclosure Statement
All authors state that they do not have any financial interests or connections, direct or indirect, that might raise the question of bias in the present work.