
Abstract
Epidermal growth factor receptor (EGFR) is one of the major molecular targets for cancer diagnosis and therapy. EGFR and EGFRvIII, mutated form of EGFR, have been identified as participating in pathogenesis of some forms of human cancers. Monoclonal antibodies (mAbs) targeting EGFR/EGFRvIII have been shown to suppress the signal transduction pathways controlling tumor cell growth, proliferation, and apoptosis. Until now, different types of mAbs or antibody fragments against EGFR family have been established. Some of these antibodies have been used clinically for treating various forms of human malignancies. More recently, a single domain antibody (sdAb) targeting this family of receptors has been introduced. The heavy chain antibodies (HCAbs) that made up variable regions of heavy chain, CH2, and CH3 domains are shown in camelids. SdAbs derived from camel HCAbs are the smallest known natural building parts for binding to antigen. They also possess a longer antigen recognizing region, which increases their capability for being more specific in target antigen enhancement. Camelid antibodies are highly valuable for their special characteristics, including heat resistance, small size, high solubility in an aqueous environment, and non-immunogenicity in a human environment. Due to these abilities, research on biotechnological production and treatment applications of recombinant smaller fragments of these only HCAbs is widely in progress. In this article, we will discuss the challenges and successes of different types of mAbs targeting EGFR/EGFRvIII in human cancer.
Epidermal Growth Factor Receptor
EGFR exists on the cell surface as inactive monomers and is activated by binding of its specific ligands. More than ten different ligands have been identified for this family of receptors (Garrett et al., 2002). Once activated, EGFR can bind with another EGFR or another member of the ErbB receptor family to create an active homo or heterodimer respectively. Binding of ligand stimulates the intrinsic protein-tyrosine kinase activity of EGFR, which initiates a signal transduction cascade leading to DNA synthesis and cell proliferation. The kinase activity can also result in autophosphorylation of tyrosine residues in the C-terminal domain of EGFR. Autophosphorylation induces downstream activation and signaling events of other proteins that are often distinct from those activated by the kinase domain of EGFR. Such proteins modulate phenotypes such as cell migration, adhesion, and proliferation (Hynes and Lane, 2005; Citri and Yarden, 2006) (Fig. 1).

Cell signaling pathways controlled by activation of EGFR. At first, binding of a ligand to EGFR results in homodimerization and phosphorylation of specific tyrosine residues in the EGFR intracellular domain. This, in turn, triggers a cascade of intracellular signals to the cytoplasm and then to the nucleus. The intracellular pathways activated by EGFR regulate gene transcription, cell-cycle progression, and proliferation, and activate a cascade of anti-apoptotic and prosurvival signals. MAPK denotes mitogen-activated protein kinase, P phosphate, PI3K phosphatidylinositol 3, 4, 5-kinase. EGFR, epidermal growth factor receptor.
Any alteration in this signaling system is thought to be involved in pathogenesis and uncontrolled growth of many common human cancers. In fact, EGFR was among the first transmembrane tyrosine kinase receptors to be directly linked with human cancer. Overexpression of growth factor receptors and their ligands, which correlates with an increase in gene amplification frequency, is observed in more than 70% of human cancers (Yarden, 2001), including lung, breast, prostate, ovary, brain, colon, bladder, thyroid, and head and neck (Gullick, 1991; Nakamura, 2007; Uberall et al., 2008; Lo et al., 2010).
Increased expression of two members of the epidermal growth factor (EGF) receptor family, EGFR and ErbB2, frequently correlates with a more aggressive clinical course of tumor. Some in vitro studies showed that inducing the expression of high levels of these two receptors in non-malignant cell lines leads to a transformed phenotype (Alroy and Yarden, 1997). Based on these observations, EGFR and its related families have been regarded as appropriate targets for cancer therapy (Yaish et al., 1998; Baselga, 2002; Cohenuram and Saif, 2007).
Mutations that lead to constant EGFR overexpression or overactivity can result in uncontrolled cell division. Several kinds of EGFR gene mutations have been identified in various types of cancer (Wikstrand et al., 1997; Kuan et al., 2001; Pedersen et al., 2001; Lorimer, 2002) from which EGFR class III mutant (EGFRvIII) is reported to be the most common variant of the EGFR identified in brain (Feldkamp et al., 1999), breast (Wikstrand et al., 1995; Ge et al., 2002), head and neck (Sok et al., 2006) ovary (Moscatello et al., 1995), prostate (Olapade-Olaopa et al., 2000), thyroid (Omidfar et al., 2009), and non-small-cell lung cancer (NSCLC) (Okamoto et al., 2003). Although EGFRvIII expression has been displayed in several cancer tissues, it is, however, weakly perceivable in normal tissues (Wikstrand et al., 1995; Feldkamp et al., 1999; Omidfar et al., 2009). From the genotype point of view, the 145-kDa EGFRvIII mutant receptor is recognized by an in-frame deletion of exons 2–7 from the extracellular region. Joining of exons 1 and 8 in mutant form creates a novel epitope with a glycine residue at the junction site, a feature that is unique to the cancer cells (Fig. 2).
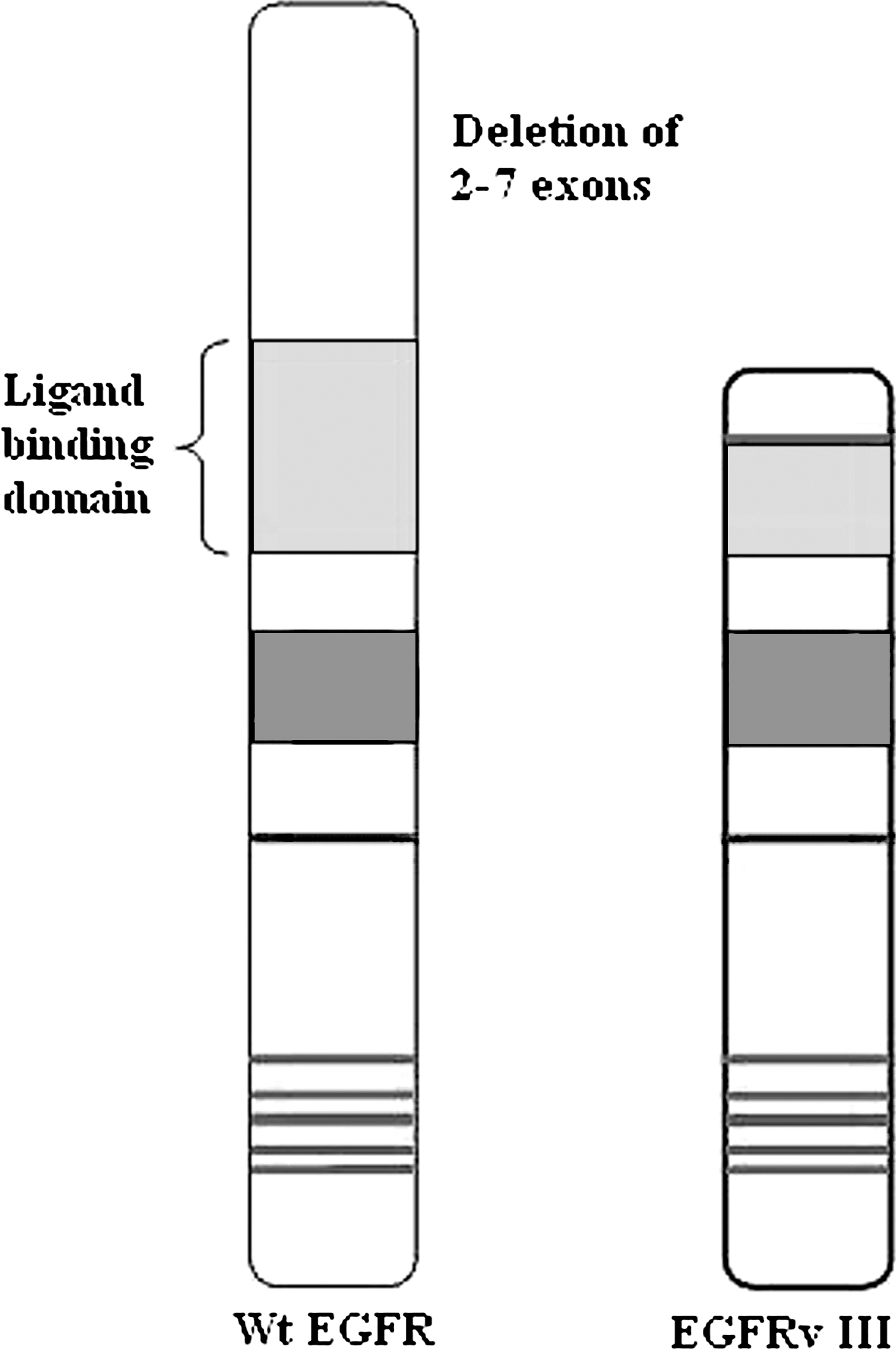
Schematic presentation of wild-type epidermal growth factor receptor and its mutant form EGFRvIII. The EGFRvIII variant receptor is produced by a deletion of exons 2–7 of the wild-type (Wt) EGFR gene. This results in an in-frame truncation of amino acids (AA) 6 to 273 in the extracellular domain of protein, yielding a constitutively active variant receptor that cannot bind to ligands. The EGFRvIII also contains a novel glycine residue inserted at the fusion junction.
EGFRvIII is not ligand dependent and, unlike EGFR, is chiefly monomeric and needs less external signals for growth and invasion (Omidfar et al., 2009). Taking all these characteristics together as well as having an exclusive cancer cells epitope at extracellular domain of EGFRvIII makes this mutant form of EGFR an extremely attractive target for cancer therapy. Scientists have generated various strategies that selectively recognize EGFRvIII over EGFR (Hills et al., 1995; Luwor et al., 2001; Mishima et al., 2001; Aldape et al., 2004; Yang et al., 2006). Finding precise ways to detect this mutation could open a window toward the targeted molecular therapy of cancers (Omidfar et al., 2009). In this article, we will focus on different types of anti-EGFR and EGFRvIII monoclonal antibodies (mAbs) that block the extracellular ligand binding domain. In addition, we will highlight therapeutic aspects of anti EGFR and mutant EGFR treatment in cancer.
EGFR Inhibition
Recent progresses in our understanding of cellular and molecular basis of human cancers have made it possible to develop new therapeutic strategies with increased efficacy and reduced toxicity. Several studies have identified cell surface antigens that are either tumor or lineage specific (e.g., CD20 in lymphoma), receptors that are overexpressed in cancer cells compared with normal cells (e.g., EGFR and HER2), or receptors that are mutated in cancer cells (e.g., mutated EGFR). Targeting these potential specific markers in cancer therapy has been the focus of many research works. The advantage of these targeted therapies is that in contrast to chemotherapy, which is very toxic and often results in harmful clinical effects, targeted therapy intends particularly at the related apostate protein. In line with this specific cancer therapy, the ErbB family of receptors was selected as the first target for cancer therapy. The reason for this was mostly because of the abundant participation of this family of receptors in many types of cancer.
In fact, EGFR is the first tyrosine kinase receptor cloned and still remains at the forefront of targeted therapies against cancer (Ciardiello and Tortora, 2008).
Historically, the first anti-EGFR inhibitors were developed in the 1980s (Masui et al., 1984; Normanno et al., 2003; Ciardiello and Tortora, 2008) and over 30 years, a large number of EGFR antagonists have been approved for the treatment of four metastatic epithelial cancers including non–small-cell carcinoma of lung, squamous-cell carcinoma of head and neck, colorectal cancer, and pancreatic cancer (Ciardiello and Tortora., 2008).
Potential therapeutic strategies that specifically target either the intracellular or extracellular segment of the EGFR/EGFRvIII and its family members include anti-receptor antibodies, small-molecular-weight tyrosine kinase inhibitors, receptor-ligand conjugates, receptor-immuno conjugates, dominant-negative receptor constructs, and antisense oligonucleotides, all of which are capable of blocking EGFR/EGFRvIII function (Panousis et al., 2005). Anti-EGFR mAbs and small-molecule EGFR tyrosine kinase inhibitors (Masui et al., 1984; Normanno et al., 2003; Ciardiello and Tortora, 2008) are two major pharmacological approaches that have been successfully tested in phase 3 trials and are now available for clinical use.
Small-molecule tyrosine kinase inhibitors, such as erlotinib and gefitinib, reversibly compete with ATP for binding to the intracellular tyrosine kinase domain of different growth factor receptors and, thereby, block the EGFR autophosphorylation and subsequent downstream signaling (Imai and Takaoka, 2006).
Antibody-based therapy appears to be the most promising approach in which the specific key points of the main pathways of cancer growth and survival are targeted. Indeed, antibodies could be used to disrupt the function of tumor cells by targeting important sites or regulators of cell proliferation, metabolism, adhesion, migration, metastasis, and other properties of malignant cells. Up to now, several specific antibodies against EGFR and its most common mutant form, EGFRvIII, have been developed. After receptor binding, these antibodies are internalized into the cell and block the cell growth (12, 25) (Fig. 3).
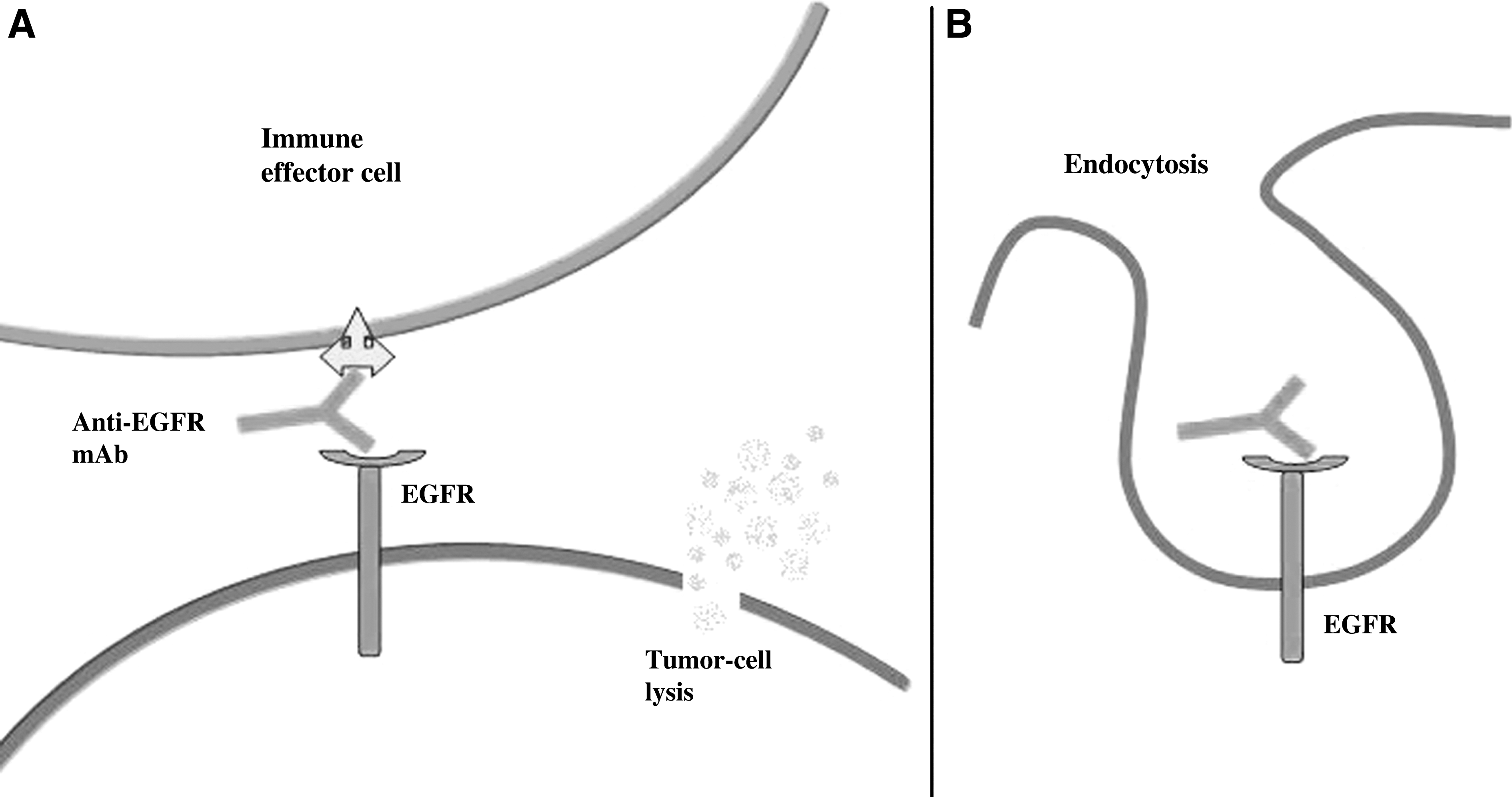
Mechanism of action of anti-EGFR mAbs in cancer cells. The anti-EGFR mAb could stimulate the host antitumor immune responses of either antibody-dependent or cell-mediated cytotoxicity
The use of antibodies to target radionuclides, drugs, toxins, and oncolytic viruses, as the next generation of mAb-based products for cancer, is also expanding. At least in case of targeting the radionucleides, clinical studies have shown that these immune-conjugates are more effective than immunotherapy with antibody alone. This highlights the enhanced efficacy achieved when a cytotoxic agent is targeted by an active antibody (Schrama et al., 2006; Oldham and Dillman, 2008).
Two Food and Drug Administration (FDA) approved radioisotope antibody conjugates are iodine-131 tositumomab (Bexxar; GlaxoSmithKline, Middlesex, United Kingdom) and yttrium-90 ibritumomab tiuxetan (Zevalin; Cell Therapeutics Inc, Seattle, WA) (Oldham and Dillman, 2008).
An approved US FDA list of anti EGFR mAbs for treatment of cancer is presented in Table 1. Some of these agents are widely used, while some agents are currently being tested for use in clinical setting. All the approved antibodies for cancer therapy are now available for treatment of human cancer. In addition to these unconjugated mAb therapies, immunoconjugate drug of gemtuzumab ozogamicin (Mylotarg; Wyeth-Ayerst, Madison, NJ) has also been approved. This calicheamicin conjugated mAb is specific to CD33 and has been used in acute myelogenous leukemia (Schrama et al., 2006; Oldham and Dillman, 2008; Deffar et al., 2009).
TheraCIM is a new humanized mAb against the human EGFR. It was developed by CIM/CIMAB/YM Biosciences and approved in China and Cuba for nasopharyngeal carcinomas; in Argentina, Colombia, and Cuba for head and neck tumors in 2005; and in Thailand (November 2010) and in Myanmar (July 2010). Clinical trials for approval by the FDA and the EMEA are ongoing. EGFR, epidermal growth factor receptor.
Conventional and Heavy Chain Antibodies
Production of antibody/immunoglobulin, a Y-shaped highly soluble multi-domain protein that circulates freely in body fluids, is one of the main actions in the vertebrate immune system. These proteins have various roles, including establishing safety and protection against foreign or non-self molecules. Antibody molecules belong to a category of proteins called glycoprotein and were first described in 1890 by Von Behring and Kitazato. The basic structural unit of the conventional antibody is made up of two identical heavy and light chains composed of constant and variable regions. The antigen binding activity of a mAb is determined by conformation of its amino acids in the complementarity determining regions (CDR). The antigen-binding site of these antibodies contains six CDR in hyper variable regions of light and heavy chain (VH) (Fig. 4).

Schematic illustration of the conventional antibodies (top) with two identical light chains and two identical heavy chains. The bottom right figure is the HCAb as it is presented in camel serum. The entire light chain (curved lines) and CH1 domain (black) are absent in HCAb. The antigen-binding domains of conventional antibodies obtained after proteolysis (Fab) or after cloning and expression of the gene VH and VL fragments are shown. A synthetic linker introduced between the VH and VL stabilizes the VH-VL dimmer and forms the scFv. The recombinant VHH, the variable domain of the HCAb, is obtained after cloning and expression induction. The IgNAR (bottom right) is a homodimer of an H-chain with five C domains and a V-NAR at its N-terminal end. The VHH is the minimal intact antigen-binding fragment that can be generated. VL, variable regions of light; VH, variable regions of heavy chain.
The antibodies are produced in two basic forms: polyclonal and monoclonal. Typically, in polyclonal antibodies, the immunological response to an antigen is heterogeneous and obtained from many different B cell repertoires, while mAbs are monospecific antibodies that are made by identical immune cells. The mAbs bind to only one specific target site on an antigen.
The first mAb was developed in 1975 using the hybridoma technique (Pandey, 2010). Over the past 30 years, a great number of mAbs have been produced and characterized against various antigens, but only a few of them have offered a clear clinical benefit in treatment of cancer (Holliger and Hudson, 2005; Schrama et al., 2006). MAbs (Kashanian et al., 2002; Paknejad et al., 2003., Omidfar et al., 2007a) are mostly used for development of clinical diagnostic tests such as enzyme immunoassays (Mohammadnejad et al., 2006), immunochromatography (Omidfar et al., 2010; Omidfar et al., 2011b, 2011c), and immunosensors (Omidfar et al., 2011a). In 1997, two decades after generation of the first mAbs, the first antibody for cancer therapy was approved by FDA (Holliger and Hudson, 2005; Schrama et al., 2006).
MAbs are typically produced in mice (murine Abs) and when used in the human body, they could be recognized as foreign particles by the immune system, provoke an allergic reaction, and rapidly be cleared from the patient's circulation. These problems enable us to have repeated antibody administration for treatment purposes (Schroff et al., 1985; Shawler et al., 1985).
Following the recent advances in genetic engineering techniques, hope for diminution of immunogenicity of the murine antibodies by producing the chimeric or humanized version of such antibodies or by development of fully human antibodies has been emerged (Boulianne et al., 1984; Morrison et al., 1984; Jones et al., 1986; Verhoeyen et al., 1988).
A chimeric antibody could be constructed using the combination of variable regions with the human constant region (Boulianne et al., 1984; Morrison et al., 1984). The humanized antibodies are fully human antibody scaffolds minus their murine antigen-binding CDRs (Jones et al., 1986; Verhoeyen et al., 1988).
The humanized or chimarized mAb have a long half-life in the blood stream, and can interact with human complement or effector cells of the patient's immune system. They behave in a manner similar to naturally occurring immunoglobulin and work along the lines of our normal antibody-based immune response as effective agents in treating patients with cancer. However, some of these engineered mAbs still have their own difficulties. For instance, the repeated use of a partially humanized mAbs in rheumatoid arthritis patients caused human-antimouse antibody responses (Smith et al., 2005).
The labeled mAbs also have therapeutic limitations due to their large molecular size that decreases their penetration to solid tumors. In addition, they stay for a long time in blood circulation and tend to accumulate in the liver (Burvenich et al., 2005; Goldenberg and Sharkey, 2007).
The continuous search for improving the antibody solid tumors penetration, reduction of blood clearance, minimizing fragment of crystallizable (Fc)-mediated side effects, and neutralizing immune responses has led to the development of smaller fragments of antibody such as Fab (one light chain and half a heavy chain), single-chain variable fragments (scFv, two variable domains, one from a light and one from a heavy chain), and, more recently, single-domain antibodies (sdAbs) (Muyldermans, 2001; Holliger and Hudson, 2005, Filpula, 2007) (Fig. 4).
Nowadays, cloning and engineering of Fab and Fv fragments have been successfully achieved in bacteria, yeast, fungi, and plants (Holliger and Hudson, 2005, Filpula, 2007), though production of scFv presents some functional limitations including aggregation due to presence of an oligopeptide linker and susceptibility of the linker to proteolytic cleavage and subsequent unfolding of the antibody construction (Muyldermans, 2001).
Recently, a novel antibody in camels (camels, llamas, and alpacas) has been discovered that is devoid of light chains and first constant domain (CH1). These heavy-chain antibodies (Fig. 4 IgG2 and IgG3 (90 KDa)) are smaller than their counterparts in conventional mammalian antibodies (150–160KDa) due to the absence of the CH1 domain (Hamers-Casterman et al., 1993) that is present in the genomic DNA, but has been removed during mRNA splicing. The variable domain of camelid heavy chain antibodies (HCAbs) consists of a single domain called VHH and constitutes the smallest known natural intact antigen binding fragment derived from a functional immunoglobulin. It has a molecular weight of only 12–15 KDa, which is even smaller than Fab fragments (∼50 kDa) and scFV (∼25 kDa). SdAbs can also be originated from the new antigen receptor antibodies (NARs) of nurse sharks (Ginglymostoma cirratum) (Greenberg et al., 1995; Bell et al., 2010).
IgNAR are also composed of two covalently associated heavy protein chains with six domains and no associated light chain (Fig. 4). Since this molecule shares several functional features with Ig isotypes, it is called IgNAR, while its N-terminal V domain is known as V-NAR.
One of the most powerful technologies developed in recent years for generating new antibody-based structures (Fab, scFV, and sdAbs) against various surface antigens is phage display technology. The major potential of this technology is the isolation of specific antibodies, proteins, and peptides from available phage libraries, which allows the selection and amplification of phage clones with specific binding activities.
Antibody fragment libraries can be constructed using Ig variable region genes derived from B cells of an immunized donor (immune library), B cells of a non-immunized donor (naive library), a combination of germline V genes and synthetic oligonucleotide sequences encoding random CDRs (semi-synthetic library), or using fully synthetic sources (synthetic library).
Biological and developmental importance of camel HCAbs is still not clearly identified; however, some functional and molecular investigations have shown that camel HCAbs have a broad range of antigenic characteristic and, thus, contribute to Ig receptor diversity (van der Linden et al., 2000; Nguyen et al., 2001; Sehrawat and SinghVeterinary, 2006). Due to the high thermostability (Omidfar et al., 2007b), production yield, detergent resistance (Arbabi Ghahroudi et al., 1997), and relatively high proteases resistance (Dumoulin et al., 2002), sdAbs derived from camel HCAbs are very suitable building parts for new antibody molecules (Revets et al., 2005). They can also be modified to obtain very high affinity either by isolation from an immune library (Li et al., 2009) or by in vitro affinity maturation (Davies and Riechmann, 1996; De Genst et al., 2004).
According to feasibility, effectiveness, and beneficial aspects of camel antibodies production, this antibody might be a more suitable substitution for the current conventional double-chained antibodies. During the last couple of years, the potential of VHHs as an attractive alternative to antigen-binding fragments from conventional antibodies such as Fabs and single chain variable fragments (scFvs) in biotechnological, diagnostic, and therapeutic applications has been demonstrated (van der Linden et al., 1999; Cortez-Retamozo et al., 2004; Stijlemans et al., 2004).
Mouse, Humanized, and Chimeric Anti-EGFR Antibodies
MAbs are among the first approaches to be investigated for cancer treatment. In the last decade, great progress in cancer therapy by mAbs has been achieved. A summary of the most important specific mAbs targeting aberrant signaling through EGFR is presented next.
Cetuximab The anti-EGFR antibody drug of cetuximab (IMC-C225, ErbituxR), a human–mouse chimeric IgG1mAb, was the first antibody approved by FDA in 2004 for the clinical treatment of metastatic cancer. This antibody binds to the extracellular domain of EGFR with a higher affinity compared with its natural ligands of EGF and transforming growth factor (TGF)-a. After binding to EGFR on the surface of cells, cetuximab promotes the internalization of receptors. This eventually blocks the ligand-binding domain and leads to a degradation of the receptors without phosphorylation and activation, a process that results in blockade of important pathways of cell division promotion. The final result would manifest as inhibition of cell growth and induction of apoptosis. Cetuximab also binds to the mutant receptor EGFRvIII, induces internalization of antibody-receptor complexes, and reduces the production of phosphorylated EGFRvIII. The safety of cetuximab alone or in combination with cytotoxic chemotherapy was evaluated in phases I, I/II cetuximab study, and in a multi-center randomized phase III study. In these trials, cetuximab was used as treatments for patients with metastatic squamous cell carcinoma of head and neck, colorectal cancer, and non-small-cell lung cancer (Pandey and Chandramohan, 2003; Martinelli et al., 2009; Wykosky et al., 2011).
Panitumumab, formerly named ABX-EGF, is the second antibody that was approved by FDA in 2006. It is a fully human G2 mAb that targets the extracellular domains of EGFR and prevents it from binding to EGF and TGF-α. This antibody has yielded exciting preclinical data, with complete eradication of established human tumors. It also presents the effective high affinity therapy (Kd=5×10−11 M) with a minimum rate of allergic reactions or anaphylaxis. IMC-225 and panitumumab share the same mechanism of action; however, given that panitumumab is a complete humanized antibody, no or little immunological response would be produced against this antibody. This property increases the efficacy and decreases the premature termination of treatment due to side effects. Panitumumab has been evaluated in clinical trials (phases I, I/II and a multi-center, randomized phase III studies) both as monotherapy and in combination with other agents for the treatment of various types of cancer, including colorectal and kidney cancer (Pandey and Chandramohan, 2003; Martinelli et al., 2009; Wykosky et al., 2011).
Matuzumab is another humanized anti-EGFR mAb (derived from the murine mAb 425) that has displayed some beneficial effects in clinical trials. A phase I study has evaluated the safety and possible advantage of combination therapy with matuzumab and a standard dose of gemcitabine. Matuzumab represses the phosphorylation process in EGFR and affects the receptor-dependent signaling and transduction (Graeven et al., 2006). This would decrease the invasion of tumor cells into healthy tissue and attenuate the propagation of tumor into new body regions.
Kamat et al., (2008) investigated the effects of chimeric mAb cetuximab and humanized mAb matuzumab combination therapy on EGFR signaling and concluded that this combination would work well in EGFR inhibition.
Nimotuzumab, also known as h-R3, is a humanized murine mAb that binds with intermediate affinity (Kd=10−8) and high specificity to the extracellular domain of EGFR. This results in blockade of ligand binding and receptor activation. In in vitro and in vivo preclinical studies, nimotuzumab revealed significant anti-proliferative, pro-apoptotic, and anti-angiogenic properties similar to those of other anti-EGFR mAbs. This antibody holds promise either as a single agent or as an adjunct to radiation therapy in phase I and II clinical trials. In clinics, it has been used for treatment of head and neck squamous cell carcinoma and glioblastoma (Boland and Bebb, 2009; Rodríguez et al., 2010).
Necitumumab is a fully-human IgG1 mAb that binds to EGFR. As on October 2009, necitumumab was started in two phase III trials as the first-line treatment for advanced non- small cell lung cancer. It has the potential benefit of lower hypersensitivity reaction risk and similar antibody-dependent cell-mediated cytotoxicity compared with cetuximab (Dienstmann and Tabernero, 2010).
Zalatumumab is a new fully humanized mAb that targets EGFR. Zalutumumab was specifically designed for the treatment of squamous cell carcinoma of the head and neck (SCCHN). The drug is in phase III trials for head and neck cancer treatment and has fast track regulatory status with the FDA for advanced metastatic SCCHN (Jeroen et al., 2008; Machiels et al., 2011).
Anti-EGFR Small Antibodies
In spite of clinical success of cetuximab and other antibody drugs for cancer treatment, their relatively large size (150 kDa) is a great restricting factor in tumor penetration (Jain et al., 2005) and in achieving a higher therapeutic index. To produce antibodies with improved tumor penetration, many different forms of antibodies have been engineered and examined. scFvs are often cleared rapidly from circulation, particularly because of their low molecular weight (MW less than 60 kDa) (Trejtnar and Laznicek, 2002). As a result, scFvs usually have a serum half life of less than 10 min (Jain et al., 2005). The efficiency of scFvs can be improved by constructing the divalent scFvs (Goel et al., 2000), tetravalent scFvs (Goel et al., 2000), and minibodies (Hu et al., 1996). However, the overall efficiency of these molecules is still less than optimal. By joining an scFv to the Fc domain, a novel antibody molecule scFv-Fc was generated. This molecule was self-amassed into a dimer and produced an Ab with a molecular weight of about 105 kDa (Wu et al., 2001). In this format, the antibody had an excellent tumor target with as high as 44% ID/g tumor uptake rate (Kenanova et al., 2007). This indicated the scFv-Fc as a potentially better antibody platform for early tumor detection than entire IgG. However, whenever an intact Fc joins with scFvs, further reduction of antibody size would be almost impossible. Indeed, smaller size antibody fragments lack the intact Fc domain and are, hence, unable to induce the antibody-dependent cellular cytotoxicity and/or complement-dependent cytotoxicity, two major mechanisms involved in the extermination of tumor tissue on antigen binding (Adams et al., 2001).
Anti-EGFR Camel Antibodies (Nanobodies)
As mentioned earlier, the new form of antibody sdAbs is known as domain antibodies or nanobodies based on whether it is derived from heavy-chain or light-chain variable regions of human antibodies (Jespers et al., 2004; To et al., 2005) or HCAbs of camels, respectively (Hamers-Casterman et al., 1993). In 2007, the generation and use of camel-derived single domain antibody fragments (VHH) directed to EGFR was reported in the therapy of non-established tumors (Roovers et al., 2007a; Roovers et al., 2007b). Some of these sdAbs were discovered to be useful for tumor imaging despite their high renal uptake (Gainkam et al., 2008; Huang et al., 2008). An estimation of the tumor-targeting capacity of the molecules using micro-positron emission tomography was also displayed (Bell et al., 2010).
In 2010, Andrea Bell et al. reported the anti- EGFR HCAb as a very promising new agent for tumor imaging and possibly cancer therapy because of better tumor penetration, higher production yield due to simpler two-chain molecular structure, lower dose requirements due to lower molecular weight, and versatility because of construction of fusions to other effectors' entities.
In 2011, Roovers et al. obtained nanobody CONAN-1 with a potent receptor antagonist property. Importantly, they have shown that in a mouse model of established A431 xenografts, this bi-paratopic nanobody format was very potent in inhibiting tumor outgrowth (Roovers et al., 2011).
In addition, two αEGFR nanobodies with pharmacokinetics and tumor uptake characteristics comparable to cetuximab and faster and deeper tumor penetration properties were constructed. Such a nanobody might be an ideal format for both imaging and therapeutic purposes (Vosjan et al., 2011).
The use of nanobodies is especially considered for identification of antigens that are usually not antigenic (hidden) for conventional antibodies. They can be an appropriate choice of antibody for economical production of antibody on a large scale.Overall, it seems that nanobodies are certainly a very favorable research device for the development of a new class of medicine (Tillib, 2011).
Anti-EGFRvIII Monoclonal Antibodies
The use of EGFR vIII as a target for cancer therapy is feasible because it expresses a high level on the surface of cancerous cells (Lorimer, 2002).
Due to the real tumor-specific nature of EGFRvIII, both polyclonal and monoclonal Abs have been developed against this mutant form of EGFR (Humphrey et al., 1990; Wikstrand et al., 1995). Several murine specific EGFRvIII mAbs with high-affinity constants were produced that could be used to assess both the quantitative and qualitative expression of EGFRvIII in biological fluids, or in cells and tissue samples derived from human tumor biopsies (Wikstrand et al., 1997).
The development of mAbs against EGFRvIII (mAb 806) and single-fragment chain constructs specific for the mutant EGFRvIII (L8A4, Y10, P14, ×32, MR1, MR1-1, 14E1) has been well described by different scientists (Reist et al., 1995; Wikstrand et al., 1995; Kuan et al., 1999; Schmidt et al., 1999; Beers et al., 2000; Kuan et al., 2000; Mishima et al., 2001). Other studies were also accomplished to investigate the structure and function of anti-EGFRvIII antibodies and their use in diagnostic and therapeutic applications (Scott et al., 2007; Sampsona et al., 2008).
MAb 806: Johns and colleagues have introduced mAb806 that identifies an unfolded region of EGFR which is only exposed when the cells either over-express EGFR or express a deletion mutant (de2-7 or vIII) of the receptor (Johns et al., 2004). MAb806 and its humanized form (ch806) have shown an impressive result in treating murine lung cancer (Li et al., 2007).
RAbDMvIII: this novel recombinant antibody specifically recognizes EGFRvIII. The specificity of this antibody for EGFRvIII and not wild-type EGFR or any other proteins was exhibited by several methods. Unlike other monoclonal anti-EGFRvIII antibodies, this antibody recognizes the EGFRvIII epitope in several assays, including ELISA, IF, IHC, western blot analysis, and flow cytometry (Gupta et al., 2010).
CH12: the ability of CH12, an anti-EGFRvIII mAb, in treatment of human hepatocellular carcinoma (HCC) was recognized by various experiments. The results demonstrated that CH12 preferentially bounds to EGFRvIII with a dissociation constant (Kd) of 1.346 nm/liter and could induce strong antibody-dependent cellular cytotoxicity (Jiang et al., 2011).
MR1-1: this is a scFv mAbs that binds with high affinity and specificity to EGFRvIII. Due to the rapid clearance and tumor uptake of scFvs, they seem a more attractive alternative to whole immunoglobulins, particularly for therapeutic and imaging applications (Shankar et al., 2006).
MAb L8A4: produced in 2010, mAb L8A4 binds specifically to EGFRvIII and is internalized quickly after receptor binding. Due to the short range of its β-emissions, labeling of this mAb with 177Lu would be an attractive procedure for treatment of tumors (Hensa et al., 2010).
Anti-EGFR vIII Camel Antibodies
Recently, a number of specific EGFRvIII sdAbs have also been constructed. In 2004, Omidfar et al. for the first time characterized and produced a new anti EGFRvIII camel sdAbs. This antibody was comparable with the murine anti-EGFRvIII antibody and had high specificity and thermostability (Omidfar et al., 2004a; Omidfar et al., 2004b). Using this HCAbs, the expression of the EGFRvIII in human neoplastic and non-neoplastic thyroid tissues was investigated by immunohistochemistry (Omidfar et al., 2009). In agreement with frequent expressions of this receptor variant in some human tumors, the study showed that EGFRvIII existed at high levels in a significant number of thyroid cancers. Though using both camel and conventional antibodies seems to be convenient for recognition of this mutant receptor, the camel antibody appeared to be more sensitive in the detection of EGFRvIII (Omidfar et al., 2009). This may be due to a longer CDR domain in HCAb, which permits a better contact with the antigens and a higher capability to identify them (Muyldermans et al., 1994).
This study was the first report of this kind that evaluated the immunohistochemical characteristics of the camel antibody for the immunohistochemical detection of EGFRvIII on thyroid tissue samples.
EG2: To increase the valence and circulating half-life of the antibody, an anti-EGFR/EGFRvIII sdAb, called EG2, was established. These constructs were examined in vitro for their kinetic binding properties to EGFR and EGFRvIII and in vivo for their pharmacokinetic characteristics and ability to target glioblastoma tumors. The results showed that EG2-hFc bounds optimally and has pharmacokinetic properties that could improve the glioblastoma targeting (Iqbal et al., 2010).
Conclusion
Size, solubility, stability, affinity, and specificity for a tumor-associated antigen are critical parameters for the successful use of antibodies in the imaging and therapy of cancer.
Due to over-expression, co-expression of the EGF receptor, its ligands, and activated mutations on the cell membranes, the increased activity of the receptor is the hallmark of many human carcinomas. Therefore, interrupting this receptor and/or the altered form became an ultimate goal of the targeted anti-cancer therapy methods.
MAbs therapy is one of the most successful pharmacological approaches that inhibits EGFR functions in cancer. Conventional antibodies of mouse and human origin are too large to have a good penetration into solid tumors, and the clearance rate is far too slow for diagnostic purposes. Therefore, size and affinity are two important parameters for the successful in vivo use of antibodies therapy.
SdAbs are an option in addition to routine IgGs and scFv fragment for imaging, diagnostic, prognosis, and therapeutic purposes. In contrast to VH domains of common antibodies, sdAbs are well expressed and shown to overcome, to a large extent, the stability, aggregation, and degradation problems often encountered with scFVs. Due to the close homology to human VH fragments, using these fragments may result in less or no immunogenicity of sdAbs in patients.