
Abstract
Metastasis is the leading cause of death in breast cancer patients. Recent evidence suggests that inflammation-related cytokine tumor necrosis factor-alpha (TNF-α) is implicated in tumor invasion and metastasis, but the mechanism of its involvement remains elusive. In this study, we employed MCF-7 breast cancer cells as an experimental model to demonstrate that TNF-α inhibits breast cancer cell adhesion and cell proliferation through hypoxia inducible factor-1alpha (HIF-1α) mediated suppression of vasodilator-stimulated phosphoprotein (VASP). We observed that TNF-α treatment attenuated the adhesion and proliferation of MCF-7 cells it also dramatically increased HIF-1α expression and decreased VASP expression. Through a variety of approaches, including promoter assay, electrophoretic mobility shift assay (EMSA), and chromatin immunoprecipitation (ChIP), we identified VASP as a direct target gene of HIF-1α. In addition, we confirmed that HIF-1α mediated the repression of VASP expression by TNF-α in MCF-7 cells. We also demonstrated that exogenous VASP expression or knockdown of HIF-1α relieved TNF-α induced inhibition of cell adhesion and proliferation. We identified a novel TNF-α/HIF-1α/VASP axis in which HIF-1α acts downstream of TNF-α to inhibit VASP expression and modulate the adhesion and proliferation of breast cancer cells. These data provide new insight into the potential anti-tumor effects of TNF-α.
Introduction
Tumor necrosis factor-α (TNF-α) is a pleiotropic proinflammatory cytokine that is produced by macrophages, neutrophils, fibroblasts, keratinocytes, natural killer, T-cells, B-cells, and even tumor cells to participate in various physiological and immune processes (Witte et al., 2007). TNF-α has been shown to have anti-tumor activity as evidenced by its cytostatic/cytotoxic effects on human tumor cell lines (Wilt et al., 1995), but recent studies have demonstrated that TNF-α also modulates matrix degradation and mediates tumor metastasis (Han et al., 2002). Moreover, TNF-α regulates the expression of cell adhesion proteins and is thus implicated in the determination of the metastatic phenotype of tumor cells (Khatib et al., 1999; Prevost-Blondel et al., 2000; Barshishat et al., 2002; Zhu et al., 2004; Chen and Geng, 2006).
A hypoxic microenvironment is crucial for tumor metastasis. Hypoxia inducible factor-1α (HIF-1α) acts as an important mediator of hypoxic response; significant associations between HIF-1α overexpression and patient mortality have been shown in cancers of the oligodendroglioma, breast, cervix, oropharynx, ovary, and endometrium (Zhong et al., 1999; Talks et al., 2000; van Diest et al., 2005). Functionally, HIF-1α activates the transcription of vascular endothelial growth factor, which is required for tumor angiogenesis, and growth factors such as insulin-like growth factor 2, to promote tumor survival. Loss of HIF-1α activity has dramatic negative effects on tumor growth, vascularization, and energy metabolism in xenograft assays, whereas the opposite effects are observed for HIF-1α overexpression (Semenza, 2002). HIF-1α activity is upregulated by pro-inflammatory messengers such as nitric oxide and pro-inflammatory cytokines including TNF-α and interleukin 1β (IL-1β) (Hellwig-Burgel et al., 1999; Albina et al., 2001). HIF-1α may provide a link between inflammatory response and hypoxia response.
Vasodilator-stimulated phosphoprotein (VASP), a member of the Ena/VASP family, is associated with the microfilament system in a wide variety of cell types where it promotes actin polymerization and assembly (Barzik et al., 2005; Schirenbeck et al., 2006). Given the crucial role of VASP in the regulation of the adherens junctions in epithelial cells (Krause et al., 2003), it is not surprising that VASP is implicated in tumorigenesis. VASP null NIH3T3 fibroblasts lost the neoplastic capabilities when injected into nude mice (Liu et al., 1999). Clinical studies have also demonstrated that VASP is overexpressed in lung adenocarcinoma (Dertsiz et al., 2005). Further, we found that the expression level of VASP increased in parallel to the pathological staging of gastric cancer, suggesting that it may regulate the invasive behavior of gastric carcinomas (Peng et al., 2009). Interestingly, by analyzing a VASP promoter in HMEC-1 cells, Rosenberger et al. (2007) revealed a transcription-mediated repression of VASP during hypoxia. Nevertheless, the functional interactions of TNF-α, HIF-1α, and VASP and the implications in tumorigenesis remain largely unexplored. In this study, we used MCF-7 breast cancer cells as a model and provided several lines of evidence to show that the TNF-α/HIF-1α/VASP axis plays a role in regulating the adhesion and the proliferation of MCF-7 breast cancer cells.
Materials and Methods
Plasmid constructs
Human VASP cDNA (I. M. A. G. E clone 824217, Proteintech group, Wuhan, China) was cloned into the pEGFP-C1 vector (Clontech, Palo Alto, CA). pCGN-HAM-HIF-1α expression vector was kindly provided by Dr. Xiao Wuhan (Institute of Hydrobiology, Chinese Academy of Sciences). The 5′ flanking region of the putative VASP promoter was amplified from human genomic DNA by PCR and cloned into pGL3-Basic vector (Promega, Madison, WI) to construct VASP reporter plasmids that spanned three different regions of the VASP promoter: −1921−+230, −518−+230, −132−+230, respectively. The putative HIF-1α binding site (5′-A/GCGTG-3′) was mutated to A/GGCCA using the previously described site-directed mutagenesis method (Rosenberger et al., 2007). All constructs were confirmed by sequencing.
Cell culture, transfection, and treatment
MCF-7 and HEK293 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), 50 U/mL penicillin-G, 50 μg streptomycin, and 1% glutamine, in a humidified atmosphere (95% air/5% CO2) at 37°C. Transient transfection was performed with Lipofectamine 2000 (Invitrogen). Cells were cultured in serum-free DMEM for at least 8 h before being treated with TNF-α (Invitrogen) at a concentration of 1 to 200 ng/mL.
Luciferase assay
MCF-7 and HEK293 cells were seeded in 24-well plates, and the next day they were co-transfected with 100 ng luciferase reporter construct, 20 ng renilla luciferase pRL-TK reporter, and 400 ng pCGN-HAM-HIF-1α. After 24 h, the cells were harvested and the luciferase activity was determined using the Dual-Luciferase™ reporter assay system (Promega). The relative light units were measured using a Glomax2020™ luminometer (Promega). Data were normalized by Renilla luciferase. Each experiment was performed at least three times in triplicate wells.
RNA interference
siRNA duplex oligonucleotides to HIF-1α (Genbank accession no. NM_001530) designed as previously described (Chen et al., 2005) were synthesized by Shanghai GenePharma, (Shanghai, China). Sequences for HIF-1α-siRNA were: 5′- CUGAUGACCAGCAACUUGAdTdT -3′. A scrambled-siRNA (5′- AGUUCAACGACCAGUAGUCdTdT-3′) was used in all experiments. The shRNA duplexes were designed against VASP (Genbank accession no. BC038224) with the following sequences: 5′-TGCTGTAAAGCATCACAGTGGCCCGGGTTTTGGCCACTGACTGACCCGGGCCAGTGATGCTTTA -3′ and were constructed into the pcDNA™6.2-GW/EmGFP vector (Invitrogen) to make pcDNA™ 6.2-GW/EmGFP-miR-VASP. Scrambled shRNA was obtained from Invitrogen and used as negative control in all experiments.
Semi-quantitative RT-PCR
Total RNA was extracted from cells with Trizol (Invitrogen). Total RNA (2 μg) was used for first-strand cDNA synthesis with RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Vilnius, LTU). Semi-quantitative PCR was performed in the presence of SYBR green using a 7500 Fast Real-Time PCR System. All PCR reactions were run in triplicate and repeated at least three times. Differences were calculated according to the ΔΔCt relative quantization method using the β-actin gene to calibrate. The primers for human VASP were 5′- AAAGTCAGCAAGCAGGAGGA -3′ and 5′- ATTCATCCTTGGGGGTTTTC -3′. The primers for human HIF-1α were 5′- GAAAGCGCAAGTCCTCAAAG -3′ and 5′- TGGGTAGGAGATGGAGATGC -3′. The primers for β-actin were 5′- CATTAAGGAGAAGCTGTGCT -3′ and 5′- GTTGAAGGTAGTTTCGTGGA -3′.
Western blotting
After transfection or TNF-α treatment for 24 h, the cells were washed with PBS and lysed in a modified RIPA buffer. Protein concentration was measured with the BCA™ protein assay kit (Pierce, Rockford, IL). Equal amounts of total protein (10 μg) were loaded, run on 10% SDS-polyacrylamide gel and transferred to PVDF membranes (Millpore, Billerica, MA). The membranes were blocked with Tris-buffered saline containing 5% nonfat milk for 2 h, then probed with VASP antibody (1:800 dilution, Alexis, San Diego, CA), HIF-1α antibody (1:1000 dilution, Abcam, Cambridge, United Kingdom), or α-tubulin antibody (1/1000 dilution, Sigma-Aldrich, St. Louis, MO) at 4°C overnight, followed by incubation with horseradish peroxidase (HRP)-linked secondary antibodies (1:5000, Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature, and detected by ECL reagents (Pierce). Experiments were repeated 3 times with similar results.
Electrophoretic mobility gel shift assay
Nuclear extracts from 293T cells were prepared with Nuclear Protein Extraction kit (Active Motif, Carlsbad, CA) following the manufacturer's instructions. Two oligonucleotides VASP promoter-wt (5′- CCCGACTTCCACTCCCGTGCGGGTGGGGGTC-3′) and VASP promoter -mut (5′- CCCGACTTCCACTCCATCACGGGTGGGGGTC -3′) were synthesized by Sangon Biotech (Shanghai, China) and then amplified by PCR. 5′ end of the sense strand was labeled with γ32P-ATP (NEN, Boston, MA) with T4-polynucleotide kinase (Promega). Nuclear extracts were incubated with 500 μg poly (dI:dC), and electrophoretic mobility shift assay (EMSA) was performed according to the protocol provided by Gel Shift Assay System (Promega).
Chromatin immunoprecipitation assay
To demonstrate direct binding of HIF-1α to the VASP promoter, SiHa cells were transfected with HIF-1α expression vector for 48 h. Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP assay kit (Millipore, Billerica, MA) according to the provided protocol. The primers used in this analysis spanned 160 bp around the putative HIF-1α binding site within the VASP promoter (5′- AGGGGCGCGACCAAATCAGT -3′ and 5′- TCATCCCAGCCCTCAGGAGG -3′). PCR products were analyzed by 2% agarose gel electrophoresis. β-actin was used as an internal control. PCR was performed for 25 cycles with the following cycling conditions: 94°C, 20 s; 68°C, 20 s; and 72°C, 20 s.
Cell proliferation assay
MTT [3-(4,5-dimethythiazoyl-2-yl)-2,5-diphenyltetrozoliumbromide] (Amresco, Solon, OH) colorimetric analysis was used to measure cell proliferation. Cells were seeded in 96-well plates at a density of 1×105/mL (100 μL per well). After 24 h the medium was removed and MTT was added to each well (20 μL of 5 mg/mL in PBS solution). The plates were incubated for 4 h at 37°C 5%CO2/95% air and then warm dimethylsulfoxide (DMSO; 200 μL) was added. Absorbance was measured at 570 nm with an ELISA plate reader (Infinite® 200 PRO, TECAN, Männedorf, CH). Three independent experiments were performed in triplicate.
Adhesion assay
Treated cells were seeded at a density of 1×105/mL (100 μL per well) in 96-well plates previously coated with fibronectin (Fn; 100 μg/mL) or laminin (Ln;7 200 μg/mL; Becton Dickinson, Heidelberg, Germany). After a 2 h incubation at 37°C in a 5% CO2 incubator, the plates were washed with PBS to remove any nonadherent cells. MTT was added to each well (20 μL of 5 mg/mL) and cells were incubated for 4 h at 37°C. DMSO (200 μL) was then added to each well, and, after shaking the plates for 10 min, the OD570 was tested. The percentage of adhesion was calculated according to the following formula: percentage of adhesion=(OD570 of cells treated/OD570 of cells untreated) ×100%.
Statistical analysis
Data were expressed as the mean±SD. Quantification of band densities was performed using the Image J. Student's t-test was utilized to compare the difference between two groups. p<0.05 was considered to be statistically significant.
Results
TNF-α inhibits adhesion and proliferation of MCF-7 cells via suppression of VASP expression
To evaluate the effect of TNF-α on breast cancer cell adhesion and cell proliferation, we treated MCF-7 cells with various doses of TNF-α. We used Fn, a high-molecular weight glycoprotein of the extracellular matrix that binds to membrane-spanning receptor proteins, and Ln, a major structural component of basement membranes, to observe cell adhesion ability. We observed that TNF-α treatment of the MCF-7 cells led to the decreased adhesion and proliferation abilities of the cells in a dose dependent manner (Fig. 1A, B). VASP is a downstream target of TNF-α signaling and has been reported to be involved in endothelial barrier function (Henes et al., 2009). We investigated the potential role of VASP in TNF-α mediated cell adhesion and proliferation by modulating VASP expression in MCF-7 cells. We observed markedly reduced adhesion and proliferation of the MCF-7 cells (Fig. 1D, E) after the specific knockdown of VASP by VASP shRNA, but not with the scrambled shRNA (Fig. 1C). In contrast, overexpression of VASP in MCF-7 cells by transient transfection of pEGFP-C1-VASP increased cell adhesion (Fig. 1D), but not cell proliferation (Fig. 1E). The inhibitory effects on cell adhesion and cell proliferation resulting from VASP deficiency could be attenuated by exogenous VASP expression (Fig. 1D, E).

TNF-α inhibits adhesion and proliferation of MCF-7 cells via the suppression of VASP expression.
We treated MCF-7 cells with TNF-α and observed that exogenous overexpression of VASP in the MCF-7 cells offset the reduced cell adhesion and cell proliferation that occur in response to TNF-α (Fig. 1F, G). MCF-7 cells depleted of VASP exhibited a lower rate of cell adhesion and cell proliferation when treated with TNF-α compared to the cells that were not treated with TNF-α (Fig. 1F, G). These data led us to hypothesize that VASP may antagonize the inhibitory effects of TNF-α on cell adhesion and cell proliferation. To test this hypothesis, we analyzed the mRNA and protein expression levels of VASP in response to TNF-α in MCF-7 cells. As expected, the expression of VASP was severely suppressed by TNF-α in a dose-dependent manner (Fig. 1H, I). Taken together, our results suggest that TNF-α attenuates MCF-7 cell adhesion and cell proliferation by suppressing VASP expression.
HIF-1α acts downstream of TNF-α to inhibit adhesion and proliferation of MCF-7 cells
TNF-α is known to regulate HIF-1α expression and to initiate its activity in human hepatoma cells (Hellwig-Burgel et al., 1999). To investigate the potential role of HIF-1α in TNF-α-induced inhibition of cell proliferation and cell adhesion, we examined mRNA and protein expression levels of HIF-1α in MCF-7 cells treated by TNF-α. The results showed that treatment of MCF-7 cells with TNF-α under normoxic conditions did not induce HIF-1α transcription (Fig. 2A), but did increase HIF-1α protein level (Fig. 2B). The increase of HIF-1 α could be due to either de novo protein synthesis or the inhibition of protein degradation; we utilized MG132, a proteasome inhibitor, and cycloheximide (CHX), a protein translation inhibitor, to distinguish between the two possibilities. In the absence of MG132 and following TNF-α stimulation, HIF-1α accumulated in the MCF-7 cells, but this accumulation decreased within 15 min after the removal of TNF-α and declined to a barely detectable level by 30 min after TNF-α removal (Fig. 2C). However, MG132 treatment did not affect TNF-α induced HIF-1α accumulation in the MCF-7 cells (Fig. 2C). These results suggest that TNF-α may inhibit the degradation of HIF-1α by the proteasome and that the prevention of protein degradation is not a significant factor that contributes to the accumulation of HIF-1α. HIF-1α protein decreased to an undetectable level in the MCF-7 cells after they were treated with TNF-α within 30 min of CHX incubation; the level of HIF-1α protein remained constant over 60 min despite the lack of ongoing protein synthesis, when the MCF-7 cells were also exposed to hypoxia mimic cobalt chloride (CoCl2; Fig. 2D). Collectively, these data suggest that HIF-1α is accumulated in MCF-7 cells upon TNF-α treatment.
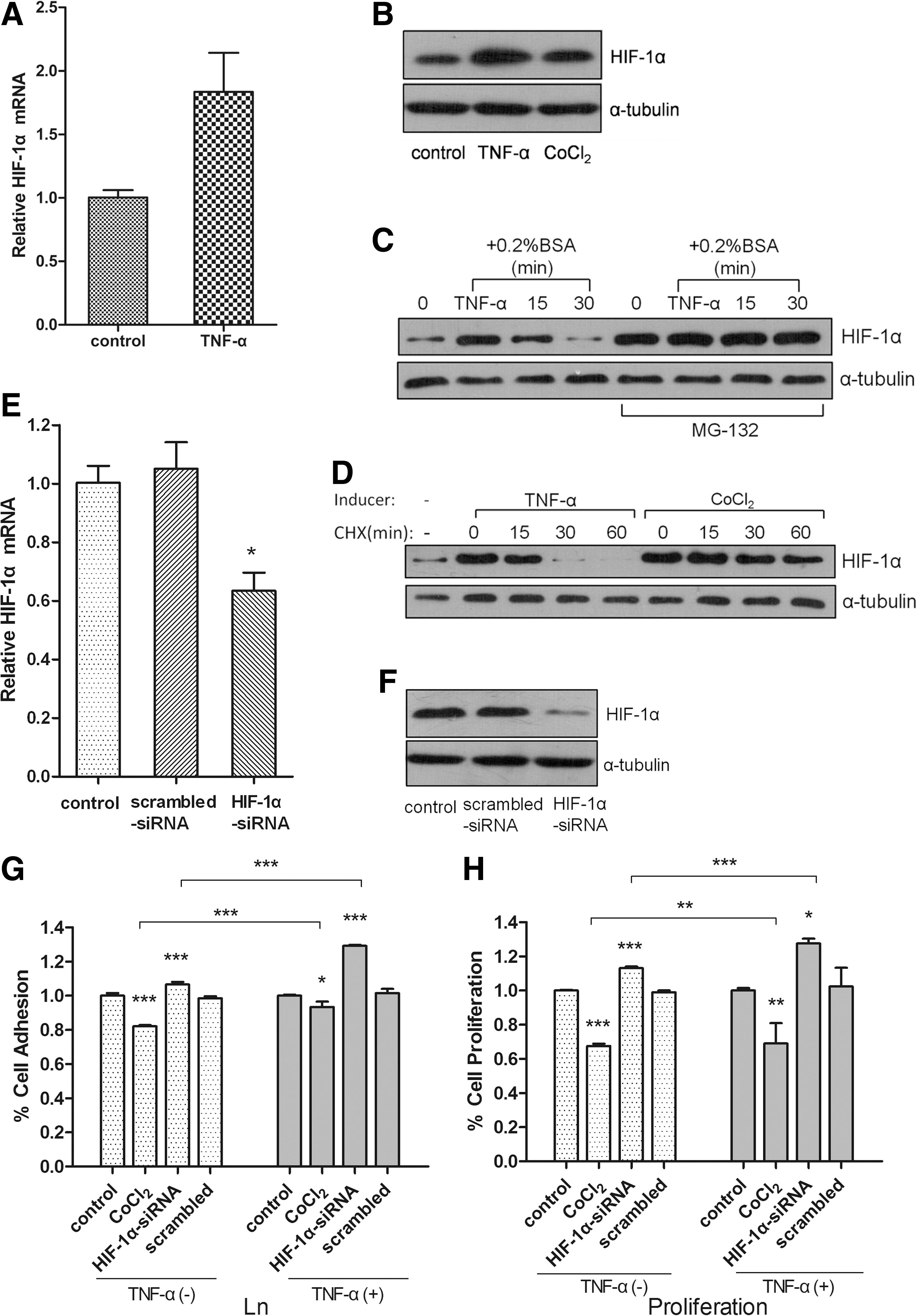
HIF-1α mediates TNF-α induced inhibition of MCF-7 cells adhesion and cell proliferation.
We observed increased cell adhesion and cell proliferation of MCF-7 cells (Fig. 2G, H) when HIF-1α was specifically knocked down by HIF-1α siRNA, but not when treated with the scrambled siRNA (Fig. 2E, F). The induction of HIF-1α by CoCl2 could significantly inhibit the adhesion and the proliferation of MCF-7 cells (Fig. 2G, H). These complementary experiments show that HIF-1α could negatively regulate the cell adhesion and cell proliferation of MCF-7 cells. Next, we assessed the effects of HIF-1α on cell adhesion and cell proliferation in the context of TNF-α treatment. The induction of HIF-1α by CoCl2 significantly augmented TNF-α-induced inhibition of cell adhesion and cell proliferation, while the knockdown of HIF-1α by siRNA significantly antagonized the TNF-α-induced inhibition (Fig. 2G, H). Therefore, our data demonstrate that HIF-1α is positively regulated by TNF-α and acts downstream of TNF-α to mediate the inhibition of adhesion and proliferation of MCF-7 cells.
HIF-1α regulates VASP expression through transcriptional repression
Because HIF-1α and VASP are both implicated in the TNF-α induced inhibition of cell adhesion and cell proliferation, we investigated the interaction between HIF-1α and VASP. Our results showed that VASP expression of mRNA levels and protein levels was repressed by HIF-1α and that VASP expression was dramatically activated by HIF-1α knockdown in MCF-7 cells (Fig. 3A, B). To confirm the negative transcriptional regulation of VASP expression by HIF-1α, we performed promoter assays. A putative promoter fragment spanning −1921−+230 at the 5′flanking sequence of the VASP gene was analyzed and the results revealed two putative HIF-1α binding sites (HBS) 5′-A/GCGTG-3′, located at −1,567 and −435 (Fig. 3C). Transient transfection of the HIF-1α expression vector and with the full promoter vector significantly inhibited VASP expression in both HEK293 cells and MCF-7 cells (Fig. 3D). However, the repressive effect disappeared when the HBS on the promoter was mutated or deleted (Fig. 3D). To further confirm that HIF-1α can regulate VASP expression directly, we performed an EMSA assay; HIF-1α bound directly to HBS consensus motif 5′-CACGTGG-3′ on the VASP promoter (Fig. 3E). ChIP analysis confirmed that HIF-1α bound the VASP promoter (Fig. 3F). Collectively, these results provide strong evidence that HIF-1α negatively regulates VASP expression through direct binding to the VASP promoter.
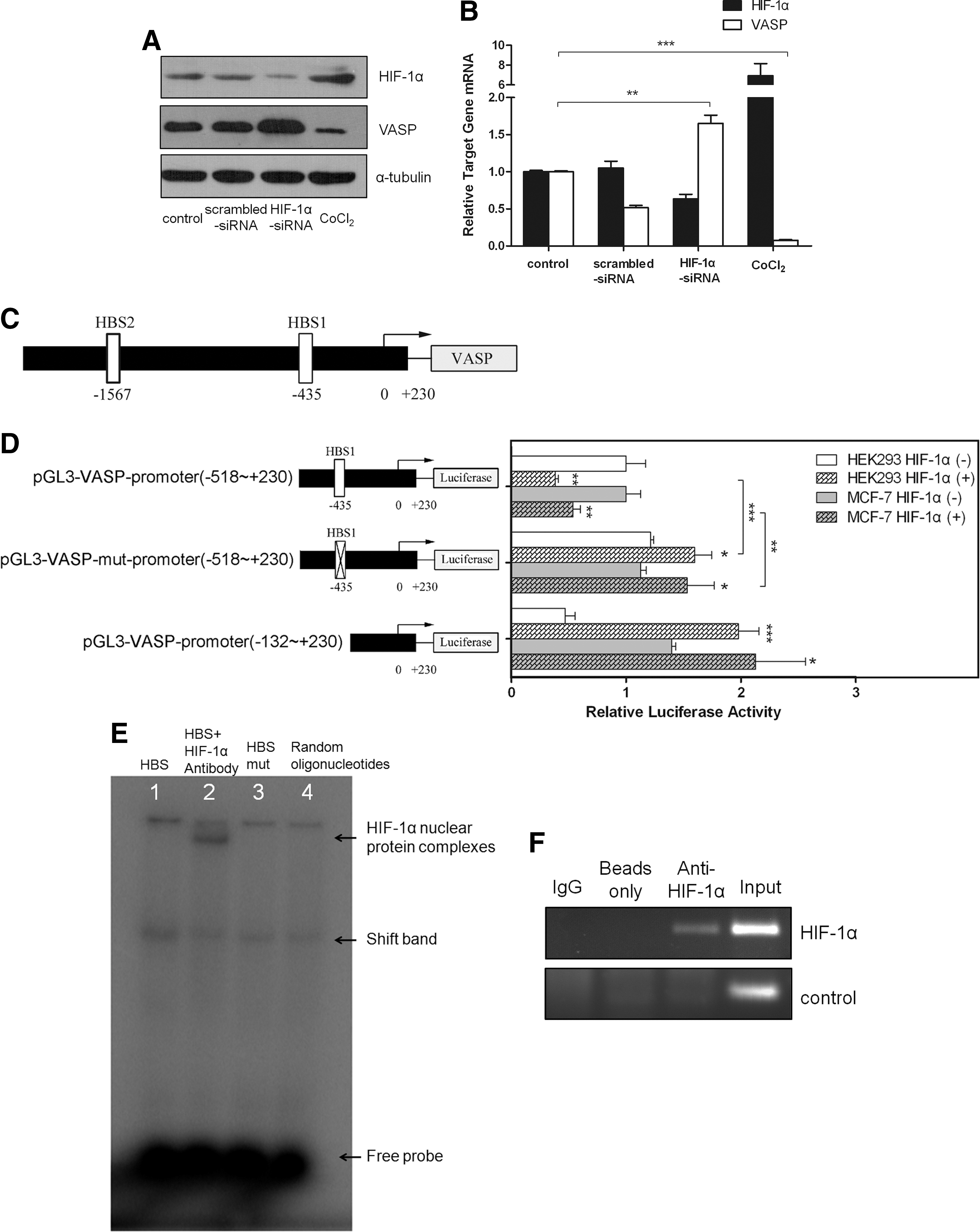
HIF-1α represses VASP expression via direct binding to its promoter.
HIF-1α mediates the repression of VASP expression by TNF-α in MCF-7 cells
To further explore whether HIF-1α is involved in TNF-α-induced suppression of VASP expression, we examined the potential role of HIF-1α in the regulation of VASP expression by TNF-α. In the absence of TNF-α, the expression of both HIF-1α and VASP was maintained at a moderate protein level and a knockdown of HIF-1α led to increased VASP expression in MCF-7 cells (Fig. 4A, B). Upon stimulation by TNF-α, the level of HIF-1α was elevated accompanied by a suppression of VASP expression. However, the suppression of VASP by TNF-α could be relieved through HIF-1α knockdown (Fig. 4A, B). These results indicate that HIF-1α mediates the repression of VASP expression by TNF-α in MCF-7 cells.
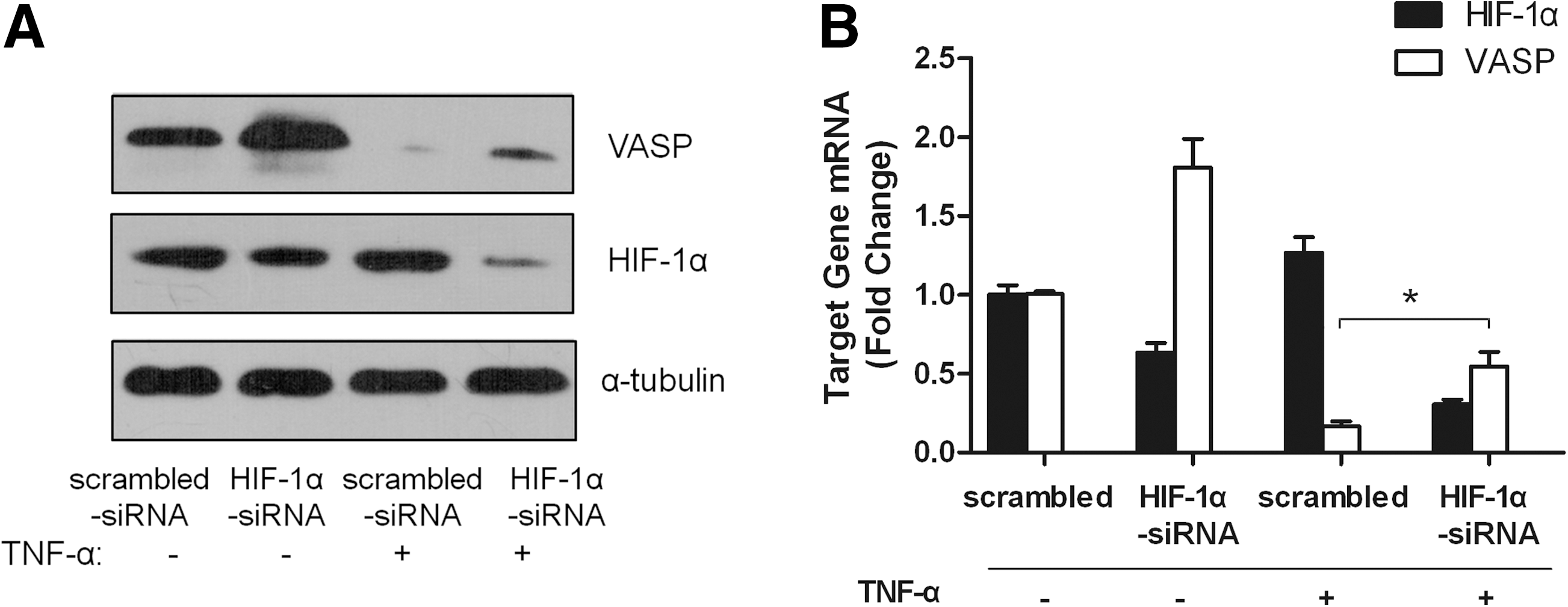
HIF-1α mediates the repression of VASP expression by TNF-α in MCF-7 cells. MCF-7 cells were transfected with HIF-1α-siRNA or scrambled siRNA for 48 h and then treated with 100 ng/mL TNF-α or untreated for 24 h as indicated.
Discussion
Numerous studies have shown that TNF-α, HIF-1α, and VASP are involved in the initiation, promotion, and progression of cancer. However, the functional interaction of these three factors and the implications of these interactions in tumor progression remain largely unexplored. For the first time, we showed that the TNF-α/HIF-1α/VASP axis plays a role in regulating the adhesion and proliferationof MCF-7 breast cancer cells. We provided a series of evidence supporting the premise that TNF-α induces the expression of HIF-1α, which in turn binds to VASP promoter and inhibits VASP transcription, consequently leading to suppressed adhesion and proliferation of MCF-7 breast cancer cells.
TNF-α is known to be involved in malignant diseases and can both inhibit and stimulate angiogenesis, depending on the applied dose. Using a disc angiogenesis system in mice and a subcutaneous osmotic millipump, it has been shown that high doses of TNF-α (1–5 μg) inhibited angiogenesis, while low doses (0.01–1 ng) stimulated angiogenesis (Fajardo et al., 1992). High doses of locally administered TNF-α even caused vessel regression and hemorrhagic necrosis, suggesting that TNF-α could be used as an anti-cancer agent (Anderson et al., 2004). However, in vivo, the secretion of endogenous TNF-α does not cause tumor regression, but rather mediates tumor progression by inducing cancer cell growth, proliferation, angiogenesis, invasion, and metastasis. These observations have been consistently shown both in animal models and in clinical studies (Mochizuki et al., 2004; Yin et al., 2009).
Recently, it was reported that TNF-α increased the attachment of melanoma cells to the ECM substrate Fn and promoted melanoma cell invasion (Katerinaki et al., 2003). Mechanistically, TNF-α stimulated cell migration and cell invasion through the activation of the transcription factor nuclear factor-kappaB and upregulation of intercellular adhesion molecule-1 (Katerinaki et al., 2006). TNF-α also promotes tumor progression in different cancer cells, in part, by stimulating MMP expression and activities at low concentrations ranging from 1–10 ng/mL (Wielockx et al., 2001). In the present study, we found that 100 ng/mL TNF-α not only inhibited MCF-7 cell proliferation but also inhibited the attachment of MCF-7 cells to Fn and Ln, suggesting that a high dose of TNF-α might inhibit tumor progression and invasion.
It has been suggested that the adhesive interaction of tumor cells with ECM components plays a critical role in the establishment of metastasis (Kato et al., 2002; Mareel and Leroy, 2003). Decreased cell-cell adhesion and increased cell-matrix adhesion are important for the detachment of metastatic cells from the primary tumor. Cell migration is a dynamic process involving polarized formation and disassembly of focal adhesions at opposite ends of the cell (Huttenlocher et al., 1995; Huttenlocher et al., 1997). In addition, adhesion dynamics must be coordinated with the assembly of new F-actin containing protrusions, which are in turn under the control of several actin nucleating and elongating molecules (Le and Carlier, 2008). Ena/VASP proteins in mammalian cells are located in focal adhesions, lamellipodia, and microspikes, and play an important role in regulating microfilament assembly (actin nucleation) and cytoskeletal organization (Sechi and Wehland, 2004). VASP binds to the proline-rich domain of the focal-adhesion proteins vinculin and zyxin (Reinhard et al., 1995; Brindle et al., 1996). Vinculin is known to contain binding sites for talin, paxillin, and F-actin, and also binds α-actinin (Brindle et al., 1996). Given their ability to interact with several cytoskeletal proteins and their localization to focal adhesions, vinculin and zyxin may act to recruit VASP and thereby initiate microfilament assembly leading to ruffling and lamellipodial extension. Consistent with these possibilities, we observed that siRNA mediated knockdown of VASP or TNF-α mediated inhibition of VASP could inhibit the proliferation ability of MCF-7 cells and inhibit MCF-7 cell adhesion to Fn and Ln, suggesting that loss of VASP function disrupts F-actin assembly and contributes to decreased cell migration ability.
Other studies also found that VASP plays an important role in the regulation of cell adhesion. A study employing VASP null mouse cardiac fibroblasts demonstrated that the adhesion of VASP-deficient cells to surfaces coated with a low concentration of Fn was delayed (Galler et al., 2006). Bear et al. (2000) reported that VASP negatively regulated cell motility. Moreover, VASP is localized to both cell-matrix adhesions and lamellipodia. Lamellipodia are the primary sites where VASP suppresses fibroblast motility (Bear et al., 2000). Theoretical and experimental analyses have shown that the effect of the cell-matrix adhesion strength on cell migration is biphasic (Huttenlocher et al., 1995). While abundant VASP is present at cell-matrix adhesions, the function of VASP in these sites has been puzzling (Zhang et al., 2006). Taken together, these studies provide more support for our findings that VASP is essential for cell adhesion. We hypothesize that VASP plays an important role in mediating the inhibitory effects of TNF-α on cell adhesion.
The focus of the current study is the characterization of signaling cascades through which TNF-α modulates the adhesion and proliferation of MCF-7 cells. We first found that TNF-α inhibited the expression of VASP in MCF-7 cells. This finding is consistent with previous studies which showed that endothelial HMEC-1 cells demonstrated a significantly reduced VASP expression level in response to TNF-α (Henes et al., 2009), and that alveolar A549 cells exhibited the repression of VASP mRNA and protein expression after stimulation with TNF-α (Henes et al., 2009). Next, we revealed that HIF-1α was positively regulated by TNF-α and that HIF-1α mediated the downstream biological effects. Rosenberger et al. (2007) described VASP as a repression target of HIF-1α, and identified one HIF-1α binding site in the VASP promoter which is coincident with our finding of one HBS site in VASP promoter as shown in Figure 3. We employed a variety of approaches including promoter assay, EMSA and ChIP to identify VASP as a direct target gene of HIF-1α. Finally, we confirmed that HIF-1α mediated the repression of VASP expression by TNF-α in MCF-7 cells.
In conclusion, our data suggest that a novel TNF-α/HIF-1α/VASP axis is crucially involved in modulating the adhesion and proliferation of breast cancer cells and provide new insight into the potential anti-tumor effects of TNF-α.
Footnotes
Acknowledgments
We thank Yihao Tian and Wuhan Xiao for advice and stimulating discussion; Guoge Han and Chen Wang for critical reading of thearticle; and Xiaoyang Wan and Daji Luo for excellent technical assistance. This study was supported by the National Natural Science Foundation of China (No. 30971132 and 81172043).
Disclosure Statement
No competing financial interests exist.