
Abstract
Gene expression is modulated by epigenetic factors that come in varying forms, such as DNA methylation, histone modifications, microRNAs, and long noncoding RNAs. Recent studies reveal that these epigenetic marks are important regulatory factors in brain function. In particular, DNA methylation dynamics are found to be essential components of epigenetic regulation in the mammalian central nervous system. In this review, we provide an overview of the literature on DNA methylation in neurodegenerative diseases, with a special focus on methylation of 5-position of cytosine base (5mC) and hydroxymethylation of 5-position of cytosine base (5hmC) in the context of neurodegeneration associated with aging and Alzheimer's disease.
Introduction
In this review, we use “epigenetic regulation” to define alterations in the structure of chromatin, a controlled complex of double-stranded DNA and histone proteins that modify access of the transcriptional machinery to a specific set of genes or chromosomal locations. So far, the molecular marks known to cause such epigenetic regulation in cells are histone modifications, DNA methylation, and hydroxymethylation. Epigenetic regulators such as histone modifications, microRNAs, and long noncoding RNAs are not discussed in this review, as excellent reviews of these can be found elsewhere (Denis et al., 2011; Kaikkonen et al., 2011; Sato et al., 2011). Combined or individually, these epigenetic marks may be potentially heritable molecular mechanisms that control gene expression.
The study of epigenetic marks has intensified in the postgenomic era, and they are emerging more and more often as mediators of gene regulation in the development of complex diseases, like cancer and neurodegenerative disorders. Epigenetic changes in postmitotic cells, such as neurons, lead to structural, physiological, and metabolic alterations, which in turn may result in subtle phenotypic differences over time, as seen, for example, in monozygotic twins (Fraga et al., 2005; Flanagan et al., 2006). One of the most studied epigenetic marks is the methylation of 5-position of cytosine base (5mC) (Fig. 1), which is dubbed “the fifth base.” Together with histone modifications, the 5mC modulates epigenetic processes, such as gene silencing via CpG islands (CGIs) (Sharma et al., 2010), and is involved in genomic imprinting (Bird, 2002; Yasukochi et al., 2010; Bartolomei and Ferguson-Smith, 2011). In addition to the fifth base, the recently rediscovered hydroxymethylation of 5-position of cytosine base (5hmC) (Fig. 1), the so-called “sixth base,” has been detected at high levels in central nervous system (CNS), though its function remains to be determined. In this review, we give a brief overview of DNA methylation in neurodegenerative diseases, with a special focus on DNA methylation dynamics within the context of neurodegeneration and its potential role in aging and Alzheimer's disease (AD).
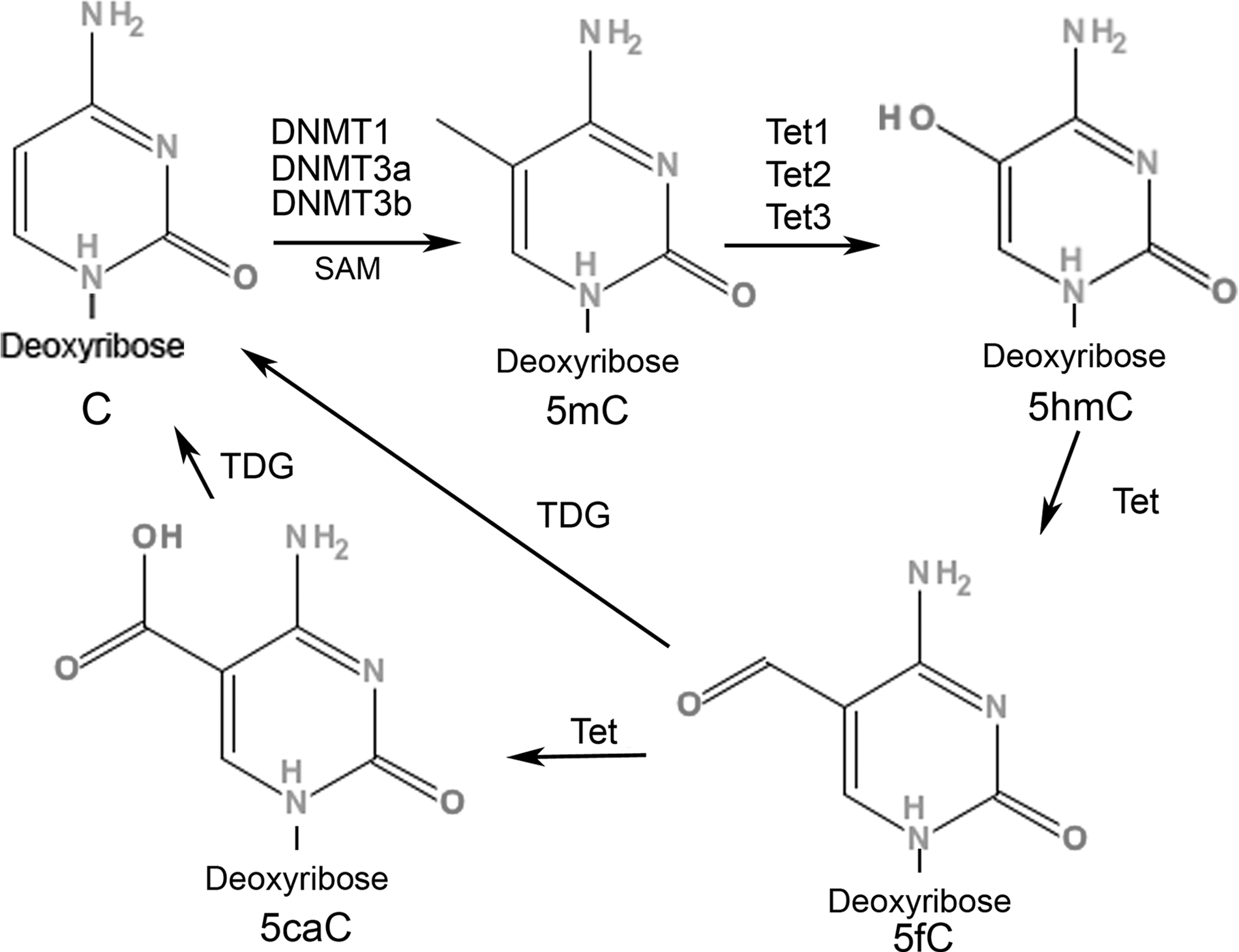
DNA methylation dynamics. Genomic cytosine nucleotide (C) is converted to 5-methylcytosine (5mC) by the addition of a methyl group from SAM (S-adenosyl-L-methionine) substrates to the 5-position C, which is catalyzed by DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b). The 5mC is further actively converted to 5-hydroxymethylcytosine (5hmC) via the ten-eleven translocation (Tet) family of enzymes (Tet1, Tet2, and Tet3) with varying affinities to 5mC in a tissue-specific manner. Oxidation of 5mC to 5hmC by Tet enzymes is proposed as an intermediate step in active DNA demethylation. Tet proteins can oxidize 5mC to 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), and 5caC can be further excised by a thymine-DNA glycosylase (TDG)–mediated base excision repair mechanism.
DNA Methylation (5mC)
In mammalian genomic DNA, methylation of cytosine is achieved by the addition of a methyl group from SAM (S-adenosyl-L-methionine) substrates to the cytosine, which is catalyzed by DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) (Fig. 1) (Bestor, 2000; Rottach et al., 2009). DNMT1 is highly expressed in neurons and shows high affinity to hemimethylated CpGs, thereby acting as a maintenance methyltransferase (Bestor, 2000). DNMT3a and DNMT3b can methylate previously unmethylated cytosines of CpG dinucleotides and thus act as de novo methyltransferases (Okano et al., 1999). Methylation of cytosine at 5-position (5mC) is considered an essential component of epigenetic regulation in mammals, as genome-wide disruption of DNA methylation is found to result in embryonic lethality (Li et al., 1992).
In mammals, the 5mC is mostly restricted to CpG dinucleotides that are confined to short genomic regions called CGIs and to promoters linked to 70% of all genes, which contain high numbers of mostly hypomethylated CGIs (Ioshikhes and Zhang, 2000; Saxonov et al., 2006). De novo methylation of CGIs is associated with gene-specific or tissue-specific gene expression regulation (Song et al., 2005; Klose and Bird, 2006) and is influenced by composition of individual cis-regulatory regions within chromatins (Weber et al., 2007). Modifications in DNA methylome have been linked to cell differentiation, reprogramming, X-chromosome inactivation, embryonic development, and cancer malignancy (Gartler and Riggs, 1983; Li et al., 1992; Hochedlinger and Jaenisch, 2006; Jones and Baylin, 2007; Portela and Esteller, 2010). Compared with histone modifications, DNA methylation is a more stable epigenetic mark, especially in differentiated cells, and is thought to be essential in gene expression control and cell type maintenance (Reik, 2007).
Recent studies show significant associations between alterations in the 5mC profile of genomic DNA and distinct phenotypes in neuronal systems. Conditional deletion of maintenance methyltransferase DNMT1 in neural precursor cells results in DNA hypomethylation in the CNS, disrupting neural control of breathing at birth, thus leading to neonatal lethality of mutant mice (Fan et al., 2001). Specific elimination of DNMT1 in murine brain causes significant DNA hypomethylation in cortical and hippocampal neurons and results in neurodegeneration that is associated with severe deficits in learning and memory behaviors, but not with motor learning and coordination (Hutnick et al., 2009). In humans, Dnmt1 mutations identified in individuals with a hereditary sensory and autonomic neuropathy type 1 (HSAN1) are linked to global hypomethylation with local hypermethylation of CGIs, potentially contributing to neurodegeneration that manifests clinically with HSAN1 (Klein et al., 2011). De novo mutations of methyl-CpG binding protein-2 (MeCP2), an X-linked gene encoding a nuclear protein that binds specifically to methylated DNA, are linked to sporadic cases of Rett syndrome (RTT), a progressive neurodevelopmental disorder in women (Amir et al., 1999). Deficiency in MeCP2 protein causes neuronal dysfunction of mature neurons in mice showing an RTT-like phenotype (Chen et al., 2001). Interestingly, while the overexpression of MeCP2 transgene in normal mice causes severe motor dysfunction, the overexpression of MeCP2 in postmitotic neurons rescues the RTT phenotype in MeCP2-mutant mice (Luikenhuis et al., 2004). Furthermore, MeCP2 regulates the expression of a wide range of genes in the hypothalamus by functioning as both an activator and a repressor of transcription (Chahrour et al., 2008). A recent study also found a strong correlation between the frequency of MeCP2 mutations and distinct phenotypes in a cohort of patients clinically diagnosed with RTT (Psoni et al., 2011).
From the studies mentioned previously, it is clear that 5mC plays a significant role in the temporal control of neural stem cell differentiation, neurodevelopment, and neurodegeneration. Additional studies show that neuronal activity alone can also cause genome-wide specific changes in the 5mC landscape. Guo et al. (2011a) demonstrated that activating dentate granular neurons of the hippocampus via electroconvulsive stimulation resulted in specific modifications in CpG methylation status, namely de novo methylation of new CpG sites and activity-induced demethylation of CpG sites that were methylated prior to stimulation. Regulation of gene expression does not seem to always directly correlate to the global levels of genomic 5mC; rather, the genomic DNA methylation characteristics of a given locus seem to contribute to whether such a locus would be subjected to specific regulatory mechanisms in gene expression. In addition to the 5mC-mediated gene regulation, the density and distribution of 5hmC in the genome show cell- and tissue-type specificity that further contributes complexity to the epigenetic regulation of gene expression.
DNA Hydroxymethylation (5hmC)
It is interesting that the initial discovery of 5hmC in the genomic DNA of mammalian brain came as a result of questioning whether the genomic DNA from somatic tissue was different from adult brain tissue due to the virtual absence of regeneration and mitosis in adult neurons (Emanuel and Chaikoff, 1960; Penn et al., 1972). Penn et al. (1972) detected 5hmC in rat brain and liver DNA preparations using a crude chromatography method, which estimated that ∼20% of total cytosine in the brain, and ∼6% in the liver, constitutes 5hmC. Decades later in 2009, Kriaucionis and Heintz, using a mass spectrometry method, confirmed the presence of 5hmC in the mammalian brain, but at a much lower 0.2% of total cytosine: about 40% as abundant as 5mC in mouse Purkinje cells (Kriaucionis and Heintz, 2009). Moreover, regardless of the age of the mouse, 5hmC was found to be 0.2% and 0.6% of total genomic DNA in granule cells and Purkinje cells, respectively (Kriaucionis and Heintz, 2009). The same year, Tahiliani et al. (2009) quantified 5hmC in the genomic DNA isolated from HEK293 cells using high-resolution mass spectrometry, providing additional evidence of the presence of 5hmC in mammalian embryonic stem cells (ESCs). In their study ∼4% of all cytosine species in CpG dinucleotides located in MspI cleavage sites (∼0.032% of all bases in the HEK293 genomic DNA) were hydroxymethylated.
Exact levels of 5hmC in genomic DNA vary among the different tissue types in different species. Even in the same species and tissue/cell type, the detected levels of 5hmC vary, and how much of this variability is due to the different detection methods used by different laboratories remains to be seen. In an attempt to determine the tissue distribution of 5hmC, Globisch et al. used an isotope-labeled derivative [D2]-hmC as a reference compound in high-performance liquid chromatography-mass spectrometry to quantify the levels of 5hmC and 5mC in parallel in genomic DNA extracted from the major organs (kidney, nasal epithelium, bladder, heart, skeletal muscle, and lung). They found that 5hmC values differed dramatically among the various tissues, in contrast to the stable amounts of 5mC. Interestingly, they also determined that the 5mC levels represent ∼4.5% of genomic DNA in the tissues examined, with the highest levels of 5hmC (0.3%–0.7%) detected in the CNS. Their findings further confirm the earlier observations that neuronal tissues contain the highest levels of 5hmC, and that these high levels are most likely related to neuronal function (Globisch et al., 2010). Knocking down DNMTs in ESCs completely eliminates 5hmC levels (Ficz et al., 2011), indicating that, at least in the ESCs, preexisting 5mC is a prerequisite for formation of all 5hmC. In mammalian ESCs the proteins responsible for active conversion of 5mC to 5hmC are the ten-eleven translocation (Tet) family of enzymes (Tet1 and Tet2) that catalyze Fe (II) and alpha-ketoglutarate (α-KG)–dependent oxidation reactions (Fig. 1) (Tahiliani et al., 2009; Ito et al., 2010).
In humans, although the levels of 5mC are comparable throughout the major organs, the levels of 5hmC again are the highest in the brain tissue (∼0.65% of all bases in genomic DNA) compared with other major organs (Li and Liu, 2011). The level of global 5hmC in normal human tissues is found to be highly variable, does not correlate with global 5mC content, and decreases as cells from normal tissue adapt to cell culture (Nestor et al., 2011). Such findings support the hypothesis that cell-specific modifications of genomic 5hmC may provide an epigenetic signature specific to a particular neuronal function or dysfunction.
Using a specific chemical-labeling method for 5hmC, our group recently generated the first genome-wide maps of 5hmC in mouse cerebellum and hippocampus during development and aging (Szulwach et al., 2011b). In this study, a detailed epigenomic view of 5hmC in CNS revealed that the global level of 5hmC in mouse brain increases with age and that terminally differentiated mature neurons show higher 5hmC content. Furthermore, 5hmC is depleted on the X chromosome of both male and female mice. Moreover, a significant increase in 5hmC densities at activated genes compared with repressed or unaltered genes during development from P7 to adult mice suggested that 5hmC is specifically acquired in developmentally activated genes (Szulwach et al., 2011b). These findings are further supported by a study from an independent group that used novel immunohistochemistry methods to detect 5hmC in mice and human tissues, which found increased levels of 5hmC in terminally differentiated neurons (Haffner et al., 2011). The presence of higher levels of 5hmC in aged brain maybe explained by the fact that oxidative stress increases with age in the brain (Floyd and Hensley, 2002), and this may contribute to the oxidation of 5mC to 5hmC (Privat and Sowers, 1996; Burdzy et al., 2002).
What Is the Function of 5hmC?
Studies show an association between the presence of 5hmC in promoters and exons with increased levels of transcription, and strong correlations between the enrichment of 5hmC and histone modifications in enhancer regions of the genomic DNA in human ESCs (Ficz et al., 2011; Song et al., 2011b; Szulwach et al., 2011a). Contrary to these findings, genome-wide mapping of Tet1 and 5hmC in ESC genomic DNA indicates that Tet1 and 5hmC are enriched in transcription start site regions with specific histone modifications known to be associated with inactive genes (Pastor et al., 2011; Wu et al., 2011). These findings overall suggest that a unique transcriptional regulation by 5hmC may contribute to the “poised” chromatin signature found at developmentally regulated genes in ESCs, rather than marking genes for active transcription.
In ESCs, the distribution of 5hmC is comparable to 5mC, and the levels of 5hmC are several-fold <5mC, except in the CGIs (Ficz et al., 2011). Although it is possible that an independent mechanism can directly hydroxymethylate the cytosine in a site-specific manner (Liutkeviciute et al., 2009), the higher levels of 5hmC in CGIs and promoter regions suggest that the Tet1 conversion of 5mC to 5hmC could be a site-specific event. These regions may be directly responsible for gene regulation at the transcriptional level. Our group has shown that, in the developing mouse hippocampus and cerebellum, tissue-specific differentially hydroxymethylated regions are located in the intragenic regions of the genome with intermediate CpG and GC content (Szulwach et al., 2011a). This finding suggests that 5hmC likely influences tissue-specific transcriptional programs during early brain development, thereby facilitating gene expression.
Factors affecting DNA methylation, such as neuronal activity, HDAC inhibitors, enriched environments (EEs), and physical exercise, are also expected to alter 5hmC levels in the brain. However, the chromosomal location and the stability of 5hmC signatures may be different. So far, it has been technically difficult to compare 5mC and 5hmC levels simultaneously in the same genomic DNA sample. Emerging methods, like new-generation chemical labeling technologies combined with single-molecule, real-time DNA sequencing (Song et al., 2011a), may provide a better understanding of 5hmC dynamics by identifying precise locations of 5hmC at a single-base resolution. Such methods may be useful for studying differentiating neurons and adult neurons undergoing oxidative stress or degeneration to identify epigenetic signatures.
Role of 5hmC in DNA Demethylation
Initially thought of as a passive replication-dependent process, demethylation of DNA is now known to occur actively in nonreplicating cells (Paroush et al., 1990; Klug et al., 2010). In the search for an active demethylase protein, Gadd45a—a growth arrest and DNA damage protein—was proposed to act as a demethylase of DNA in both proliferating and nondividing cells, and thus promote DNA repair (Barreto et al., 2007). However, neither global nor locus-specific methylation increases were observed in Gadd45a−/− mice (Engel et al., 2009), and the DNA demethylation activity of Gadd45a at a genome-wide level in mammals has not been confirmed (Jin et al., 2008). However, a more recent study found that Gadd45b, another member of the Gadd45 family of proteins, is an essential protein factor for activity-dependent demethylation of CpG regions in the promoter of genes involved in adult neurogenesis (Ma et al., 2009). Given that global DNA demethylation is a continuous event in somatic cell differentiation and requires rapid DNA replication (Shearstone et al., 2011), more studies are needed to uncover additional protein factors already involved in passive DNA demethylation in somatic cells and active DNA demethylation in nonreplicating cells, such as adult neurons.
Oxidation of 5mC to 5hmC by Tet enzymes is also proposed as an intermediate step in active DNA demethylation. A possible role for 5hmC in the DNA demethylation process in mammalian cells comes from studies showing that Tet proteins can oxidize 5mC to 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), and that 5caC can be excised further by a thymine-DNA glycosylase (TDG)–mediated base excision repair mechanism (Fig. 1) (Tahiliani et al., 2009; Guo et al., 2011b; He et al., 2011; Ito et al., 2011). In mouse preimplant zygotes, an increase in 5hmC is associated with a decrease in 5mC, suggesting that 5mC is actively converted to 5hmC by Tet3, which is the only isoform of Tet proteins expressed in zygotes (Iqbal et al., 2011). Moreover, the 5hmC mark associated with the paternal genome in zygote is gradually lost in a DNA replication–dependent manner during preimplantation zygote development (Inoue and Zhang, 2011). It remains to be seen whether such active and passive mechanisms of 5hmC-mediated DNA demethylation occur in postmitotic cells, such as terminally differentiated adult neurons.
DNA Methylation in Aging and AD
At a cellular level, aging is characterized by oxidative stress, disturbed calcium homeostasis, chromosomal instability, impaired DNA repair, and the accumulation of nuclear and mitochondrial DNA damage (Chouliaras et al., 2010). Combined or individually, these factors can lead to neuronal aging, neurodegeneration, and cell death in the central nervous system, potentially contributing to the development of age-related dementia and AD, with the aging being the highest risk factor for AD (Blennow et al., 2006), which is a progressive dementia that manifests itself as a progressive impairment in memory, judgment, decision-making, orientation to physical surroundings, and language. The number of people affected by age-related dementia is estimated to double every 20 years, and should reach 81.1 million by 2040 (Ferri et al., 2005). Genetic association studies have identified rare, fully penetrant mutations in three genes: amyloid-protein precursor (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). These mutations are responsible for familial early onset AD, while the majority of AD cases (∼95%) are nonfamilial, late-onset (>65 years) sporadic forms (LOAD) that have no clear genetic association (Hollingworth et al., 2011).
The role of epigenetics in aging and related neurological disorders has become a focus of inquiry with the discovery that global cytosine methylation of genomic DNA decreases with age in various tissues of mammals (Vanyushin et al., 1973; Wilson et al., 1987; Fuke et al., 2004). A recent study demonstrated that immunoreactivity of 5mC in the neurons of postmortem cortical tissue from AD patients is significantly less than in age-matched controls, and the levels of 5mC inversely correlate with the markers of late-stage neurofibrillary tangles in the same neurons, suggesting that a significant global loss of 5mC takes place in the AD brain (Mastroeni et al., 2010). However, a low-CpG density region of APOE ɛ4 promoter, which is a high-risk marker gene associated with LOAD, is found to be hypermethylated in LOAD patients (Corder et al., 1993; Wang et al., 2008). These findings illustrate the multifaceted role of DNA methylation in the neurodegenerative process.
The 5hmC has also been implicated in age-related neurodegeneration. A recent gene ontology pathway analysis of over 5000 genes that acquired 5hmC during aging identified a significant enrichment of pathways associated with age-related neurodegenerative disorders (Song et al., 2011b). Determining the AD-specific acquisition of 5hmC or any AD-specific alterations in 5hmC levels (or genomic distribution) may reveal specific loci that are most affected in AD patients. It is possible that distinct modifications on the 5hmC genomic landscape may provide an effective biomarker that can be used to screen for AD risk.
There have been extensive studies examining the physiological and behavioral effects of environmental factors on aging and AD animal models [reviewed in (Redolat and Mesa-Gresa, 2011)]. For example, an EE and voluntary exercise are found to improve learning and memory, to increase neurogenesis and angiogenesis in the hippocampus of aged mice (Kempermann et al., 1997; Nilsson et al., 1999; van Praag et al., 2005; Harburger et al., 2007), and to potentially slow the progress of brain aging in rodents (Leggio et al., 2005; Qiu et al., 2011). In addition to improving health and cognitive function, voluntary physical exercise can also delay the cognitive deterioration commonly observed in AD patients (Cotman et al., 2007; Rolland et al., 2008). Indeed, a recent study showed that, in a mouse model of spinocerebellar ataxia type 1 (SCA), which is a fatal neurodegenerative disease caused by expansion of polyglutamine in Ataxin-1 (ATXN1) in humans, even a mild exercise regimen can mitigate the disease phenotype (Fryer et al., 2011). However, the epigenetic changes caused by such environmental factors on CNS are not well known. Characterization of 5mC and 5hmC marks in the CNS of laboratory animals, as well as in primates that are exposed to such environmental factors, may answer some of the questions relating to the molecular mechanism behind the effects of EE and physical exercise in aging (Fig. 2). A recent study showed that early-life exposure to lead (Pb) in primates alters the expression of genes with CpG-rich 5′-untranslated regions, while also decreasing the expression of DNMT1, DNMT3a, and MeCP2 (Bihaqi et al., 2011), suggesting that an environmental factor can specifically alter the DNA methylation state of genes, thereby regulating their expression as a response to the specific factor in the environment. It is still not known whether activity-induced changes in global 5hmC levels or its genomic distribution are reversible once such factors are eliminated from the environment.
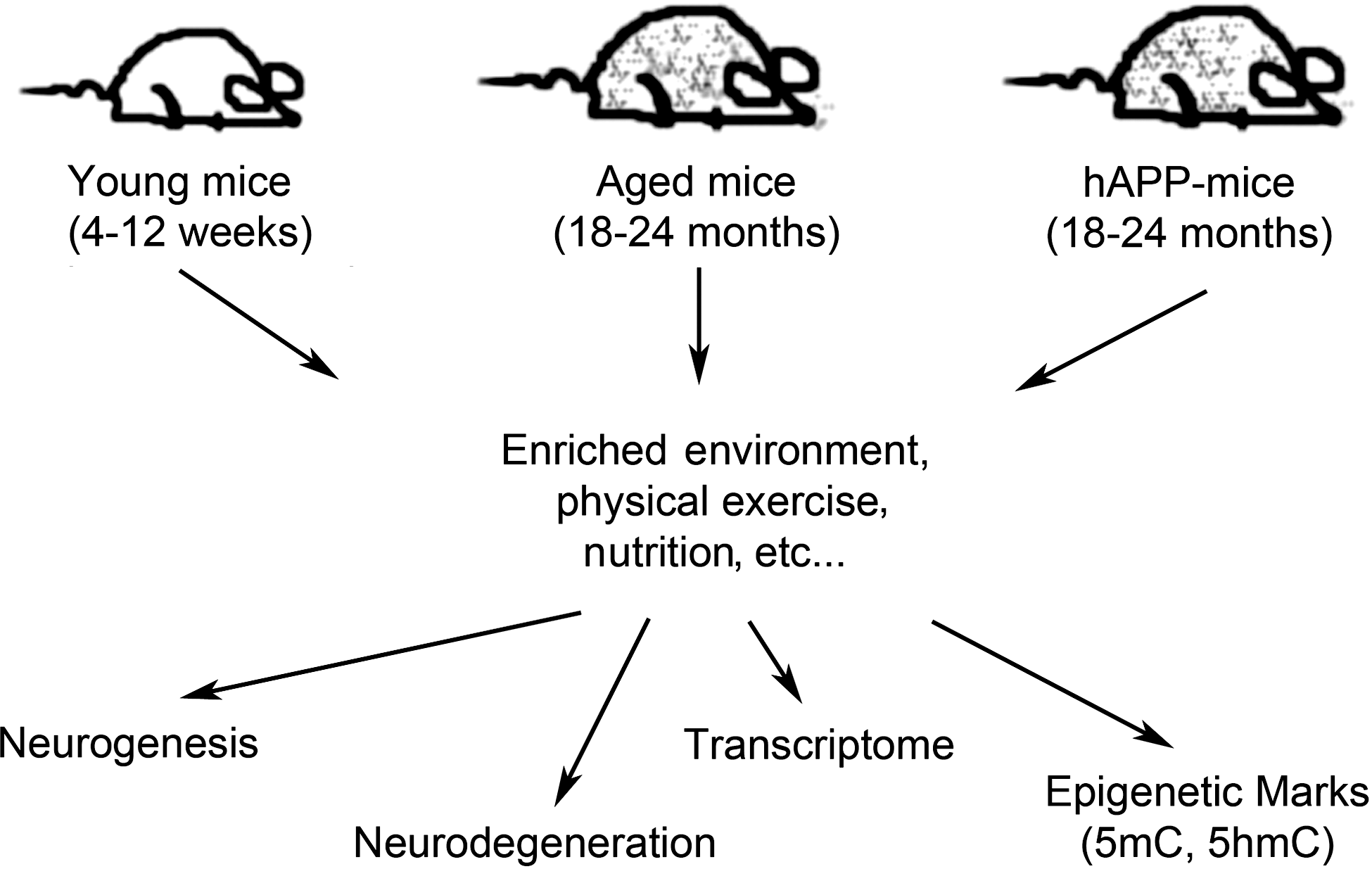
Finding age-specific epigenetic marks altered by environmental factors. Exposing young and aged mice, as well as established neurodegenerative disease models (e.g., hAPP mice), to defined environmental factors, such as an enriched environment, nutrition, and physical exercise, may alter the epigenetic landscape of genomic DNA. Subsequent examinations of age-susceptible brain regions of these mice (e.g., hippocampus, cortex, and cerebellum) for aberrant neurodegeneration and/or neurogenesis that show signature 5mC and 5hmC profiles may reveal the chromosomal loci responsive to such environmental factors.
Similarly, studies looking into the effects of nutrients and pharmacological agents on global DNA methylation are promising but inconclusive or limited in their scope in terms of genome-wide characterization of DNA methylation (Choi et al., 2005; Harris et al., 2011; Schiepers et al., 2011; Zhang et al., 2011). A genome-wide analysis of 5mC and 5hmC marks is needed to decipher the roles of specific environmental factors on the aging process, as well as age-related neurodegenerative disorders, such as AD.
Concluding Remarks
Unlike the genome of an organism, the epigenome is largely malleable and reactive to environmental factors. The molecular dynamics of 5mC and 5hmC illustrate this point clearly. In addition to histone modifications and long noncoding RNAs, which also play an integral part in the epigenetic regulation of gene expression, the high levels of 5mC and 5hmC in the CNS further highlight the functional importance of these epigenetic marks. Age- and cell-specific modifications of 5hmC in postmitotic neurons and their distinct intergenic distributions in ESCs make the 5hmC a specific epigenetic mark and an ideal biomarker for predicting the onset of neurodegenerative disorders, with or without the underlying genetic mutations. Understanding the epigenetic dynamics of Alzheimer's disease and age-related dementia would give us deeper insight into the disease etiology, aid in intervention and management of disease progression, and most importantly provide novel targets for pharmacological interventions.
Footnotes
Acknowledgments
The authors would like to thank C. Strauss for critical reading of the manuscript. H.I. is supported by the Training Program in Human Disease Genetics funded by NIH (T32MH087977). P.J. is supported by NIH grants (NS051630, MH076090, and P50AG025688).
Disclosure Statement
No competing financial interests exist.