
Abstract
The mitochondrial genome (mitogenome) of Leucoptera malifoliella (=L. scitella) (Lepidoptera: Lyonetiidae) was sequenced. The size was 15,646 bp with gene content and order the same as those of other lepidopterans. The nucleotide composition of L. malifoliella mitogenome is highly A+T biased (82.57%), ranked just below Coreana raphaelis (82.66%) (Lepidoptera: Lycaenidae). All protein-coding genes (PCGs) start with the typical ATN codon except for the cox1 gene, which uses CGA as the initiation codon. Nine PCGs have the common stop codon TAA, four PCGs have the common stop codon T as incomplete stop codons, and nad4l and nad6 have TAG as the stop codon. Cloverleaf secondary structures were inferred for 22 tRNA genes, but trnS1(AGN) was found to lack the DHU stem. The secondary structure of rrnL and rrnS is generally similar to other lepidopterans but with some minor differences. The A+T-rich region includes the motif ATAGA, but the poly (T) stretch is replaced by a stem-loop structure, which may have a similar function to the poly (T) stretch. Finally, there are three long repeat (154 bp) sequences followed by one short repeat (56 bp) with four (TA)n intervals, and a 10-bp poly-A is present upstream of trnM. Phylogenetic analysis shows that the position of Yponomeutoidea, as represented by L. malifoliella, is the same as traditional classifications. Yponomeutoidea is the sister to the other lepidopteran superfamilies covered in the present study.
Introduction
The Lepidoptera includes moths and butterflies, with more than 160,000 described species distributed worldwide in 124 families (Kristensen et al., 2007). Leucoptera malifoliella Costa belongs to Lyonetiidae (Lepidoptera). Its junior synonym is Leucoptera scitella Zeller (Mey, 1994). Lyonetiidae is a microlepidopteran family with more than 500 described species. These moths are small and slender, with a very narrow forewing and a wingspan, which rarely exceeds 1 cm. Their larvae are generally leaf miners. L. malifoliella damage apples, pears, and other plants, and withering fruit tree leaves. Recent studies focused on the sex pheromone for prevention and control (Francke et al., 1987; Koutinkova et al., 1999), but few studies have been carried out on their mitogenome, and there is a lack of mitogenome data.
In this study, we described the complete mitogenome of L. malifoliella, the first sequenced specie of Lyonetiidae, and compared its features with other available lepidopteran mitogenomes. Finally, the phylogenetic relationships among the lepidopteran superfamilies were reconstructed using complete mitochondrial genomes.
Materials and Methods
DNA sample extraction
Larvae were collected from an orchard in Beijing, China, L. malifoliella were identified according to Mey (1994), and raised in laboratory. The hatched moths were collected, preserved in 100% ethanol, and stored at −20°C. Total DNA was extracted and isolated from single specimens using the DNeasy Tissue kit (QIAGEN) according to the manufacturer's instructions.
Primer design, polymerase chain reaction, and sequencing
Short fragment amplifications were performed using the universal polymerase chain reaction (PCR) primers from Simon et al. (1994). The degenerate and specific primer pairs were designed based on the known mitochondrial sequences in Lepidoptera, or designed by Primer5.0 software on the fragments that we previously sequenced (Table 1). All the primers were synthesized by Shanghai Sangon Biotechnology Co., Ltd. (Beijing, China). For fragments of length less than 2 kb, PCR conditions were as follows: 95°C for 5 min; 34 cycles of 94°C for 30 s; 50°C–55°C (depending on primer combinations), 1–3 min (depending on putative length of the fragments) at 68°C; and a final extension step of 72°C for 10 min. For fragments of length more than 2 kb, PCR conditions were as follows: 92°C for 2 min; 40 cycles of 92°C for 30 s, 50°C–55°C for 30 s (depending on primer combinations), 60°C for 12 min; and a final extension step of 60°C for 20 min.
Primers newly designed for this genome.
Primers from Lee et al. (2006).
Primers from Zhao et al. (2010).
Primers from Simon et al. (1994).
The entire mitogenome of L. malifoliella was amplified in 14 fragments. For most fragments, we used 2× Taq PCR MasterMix (Tiangen Biotech Co., Ltd., Beijing, China) in the amplification; for fragments longer than 2 kb (cox2-nad5 and nad5-cob) and with higher AT contents (A+T-rich region), amplification used Takara LA Taq (Takara Co., Dalian, China). All amplifications were performed on an Eppendorf Mastercycler and Mastercycler gradient in 50-μL reaction volumes. The reaction volume of 2× Taq PCR MasterMix contains 22 μL sterilized distilled water, 25 μL 2× Master Mix, 1 μL of each primer (10 μM), and 1 μL of DNA template; the one of Takara LA Taq consists of 26.5 μL of sterilized distilled water, 5 μL of 10× LA PCR Buffer II (Takara), 5 μL of 25 mM MgCl2, 8 μL of dNTPs Mixture, 2 μL of each primer (10 μM), 1 μL of DNA template, and 0.5 μL (1.25 U) of TaKaRa LA Taq polymerase (Takara).
The PCR products were detected via electrophoresis in 1% agarose gel, purified using the 3S Spin PCR Product Purification Kit, and sequenced directly with ABI-377 automatic DNA sequencer. All fragments were sequenced from both strands. Short amplified products were sequenced directly by internal primers, long amplified products were sequenced completely by primer walking, but the rrnS-nad2 regions were sequenced after cloning. The purified PCR products were ligated to the pEASY-T3 Cloning Vector (Beijing TransGen Biotech Co., Ltd., Beijing, China), and then sequenced by M13-F and M13-R primers and walking. Sequencing was performed using ABI BigDye ver 3.1 dye terminator sequencing technology and run on ABI PRISM 3730×1 capillary sequencers.
Analysis and annotation
Sequence annotation was performed using the DNAStar package (DNAStar Inc. Madison, WI). The tRNA genes were identified using the tRNAscan-SE v.1.21 software (Lowe and Eddy, 1997). The putative tRNAs were then confirmed by sequence alignment with other insects of lepidoptera using the Bioedit (Hall, 1999). Secondary structure was inferred using DNA-SIS 2.5 (Hitachi Engineering, Tokyo, Japan). The trnS1(AGN) secondary structure was developed as proposed by Steinberg and Cedergren (1994). rrnL and rrnS secondary structures were drawn by XRNA (developed by B. Weiser and available at
Phylogenetic analysis
To infer the phylogenetic relationships of lepidopterans, other available complete mitogenomes in Lepidoptera were obtained from GenBank. Bactrocera oleae (NC_005333) (Nardi et al., 2003) and Anopheles gambiae (NC_002084) (Beard et al., 1993) were used as outgroups. The alignment of the amino acid sequences and nucleotide sequences of each of the 13 mitochondrial PCGs was performed with MUSCLE (Edgar, 2004) using default settings, and concatenated into an amino acid (3,872 sites in length) and nucleotide (11,616 sites in length) matrix. The concatenated set of amino acid sequences and nucleotide sequences were used in phylogenetic analyses, using Bayesian Inference (BI) and Maximum Likelihood (ML) methods. Substitution model selection was conducted via a comparison of Akaike Information Criterion scores (Akaike, 1974), calculated using the programs ProTest ver. 1.4 (Abascal et al., 2005) for amino acid sequence alignment and Modeltest ver. 3. 7 (Posada and Crandall, 1998) for nucleotide sequence alignment. The MtRev (Adachi and Hasegama, 1996)+I+G model and GTR (Lanave et al., 1984)+I+G model were chosen as the best-fitting model for amino acid sequences and nucleotide sequences, respectively, for BI analyses and ML analyses. The BI analysis was conducted using MrBayes 3.1 (Huelsenbeck and Ronquist, 2001) with four independent Markov chains run for 1,000,000 metropolis-coupled MCMC generations, with tree sampling every 100 generations and a burn-in of 2000 trees. The ML analysis was performed using RAxML (Stamatakis, 2006) with 1000 bootstrap replicates.
Results and Discussion
Genome structure and organization
The L. malifoliella mitogenome is a circular molecule of 15,646 bp in length, deposited in GenBank under accession number JN790955. The L. malifoliella mitogenome showed the typical metazoan gene content, containing 13 PCGs, 2 rRNAs, 22 tRNAs, and noncoding regions. The gene order in L. malifoliella is A+T-rich region-trnM-trnI-trnQ, whereas the ancestral gene order for the Lepidoptera is A+T-rich region-trnI-trnQ-trnM (Junqueira et al., 2004). This placement of trnM may be a molecular feature exclusive to lepidopteran mitogenomes (Cameron and Whiting, 2008).
The L. malifoliella mitogenome is biased toward A+T (82.57%) with the value falling into lepidopteran range of 77.84% (Ochrogaster lunifer, Salvato et al., 2008) to 82.66% (Coreana raphaelis, Kim et al., 2006). The A+T content was 80.24% in PCGs, 85.49%, in rrnL genes, 87.14% in rrns genes, and 95.36% in the A+T-rich region. These values were also high in other lepidopterans reported (Table 2).
Protein-coding genes
The initial and termination codons of 13 PCGs are shown in Table 3. Twelve PCGs start with a typical ATN codon (ATT for nad2, nad3, nad5, atp8; ATA for cox2, nad6, nad1; ATG for atp6, cox3, nad4, nad4l, cob). The exception is the cox1 gene, which uses CGA as the start codon. Seven PCGs have the common stop codon TAA, nad4l and nad6 have the common stop codon TAG, and four PCGs have the codon T as incomplete stop codons, which was also found in other animal mitochondrial genes (Clary and Wolstenholme, 1985).
The putative start codons of PCGs in the L. malifoliella mitogenome are ATN, except for the CGA start codon of the cox1 gene. The start codon of the cox1 gene is controversial in many studies. The putative codon CGA is common across insects (Anabrus simplex, Fenn et al., 2007; Adoxophyes hnmai, Lee et al., 2006; Manduca sexta, Cameron and Whiting, 2008; O. lunifer, Salvato et al., 2008; Eriogyna pyretorum, Jiang et al., 2009; Phthonandria atrilineata, Yang et al., 2009; Hyphantria cunea, Liao et al., 2010; Artogeia melete, Hong et al., 2009; Antheraea yamamai, Kim SR et al., 2009; Eumenis autonoe, Kim et al., 2010). The tetranucleotides TTAG and hexanucleotide TATTAG have also been proposed as start codons for the cox1 gene (Parnassius bremeri, Kim et al., 2009; C. raphaelis, Kim et al., 2006; Antheraea pernyi, Liu et al., 2008; Ostrinia nubilalis, Ostrinia furnacalis, Coates et al., 2005; Bombyx mandarina, Yukuhiroetal., 2002; Papilio xuthus, Feng et al., 2010). However, TTAG lacks absolute conservation and may serve alternative functions, not always as an initiation codon. Alignment of the mitogenome sequence from all Lepidopterans had shown that an arginine (CGR) functions as the start codon for the cox1 gene (Margam et al., 2011). In our study the start codon of the cox1 gene is CGA according to the alignment of the lepidoterans (Fig. 1).

Alignment result of trnY and cox1 in 34 Lepidopterans. The dotted line and underline indicate the locations of cox1 and trnY, respectively. The overlapping base between trnY and cox1 is marked gray.
Transfer and ribosomal RNA genes
The 22 tRNA genes ranged from 63 to 75 nucleotides. Fourteen tRNAs are coded on the J-strand and 8 on the N-strand, as with other Lepidoptera. The trnK anticodon is TTT, which is unusual in this insect order. Complete cloverleaf secondary structures could be inferred for 21 of the 22 tRNAs. The secondary structure of trnS1(AGN) was incomplete, lacking the DHU arm (Fig. 2). A total of 43 unmatched base pairs were scattered throughout the 21 tRNA genes, including 15 pairs in the DHU stems, 11 pairs in the amino acid acceptor stems, 9 pairs in the TΨC stems, and 8 pairs in the anticodon stems. Twenty-one of these are G-U pairs, which form a stable hydrogen-bonded pair. The remaining were C-A, C-U, G-G, G-A, and U-U mismatches.

Putative secondary structures for the tRNA genes of Leucoptera malifoliella mitogenome.
As in the other insect mitogenome sequences, two rRNA genes were present in L. malifoliella. The rrnL gene (1351 bp) was found between trnL(CUN) and trnV, and the rrnS (770 bp) between trnV and the A+T-rich region. Both the secondary structure of rrnL and rrnS conform to the models proposed for other insects (Cameron and Whiting, 2008; Wei et al., 2009; Wei et al., 2010). Forty-nine helices are present in rrnL of L. malifoliella, as in G. molesta (Gong et al., 2011), M. sexta (Cameron and Whiting, 2008), Drosophila melanogaster (Schnare et al., 1996), and Apis mellifera (Gillespie et al., 2006). There is a large internal loop among H991, H1057, and H1087, which is similar to G. molesta, and differs from M. sexta. The microsatellite sequence of (TA)n inserted in the loop region of H2347 in Adoxophyes honmai (Lee et al., 2006 ), Spilonota lechriaspis (Zhao et al., 2010), G. molesta, which belong to Tortricidae, is not present in L. malifoliella (Fig. 3). Twenty-nine helices present in rrnS of L. malifoliella belong to three domains, as in G. molesta, M. sexta, and A. mellifera. The structures of Helix H47, H673, H1303, H1047, H1068, H1074, and H1113 are different from M. sexta, but similar to G. molesta, with the exception of H47, which has a shorter loop length in L. malifoliella compared to G. molesta (Fig. 4).
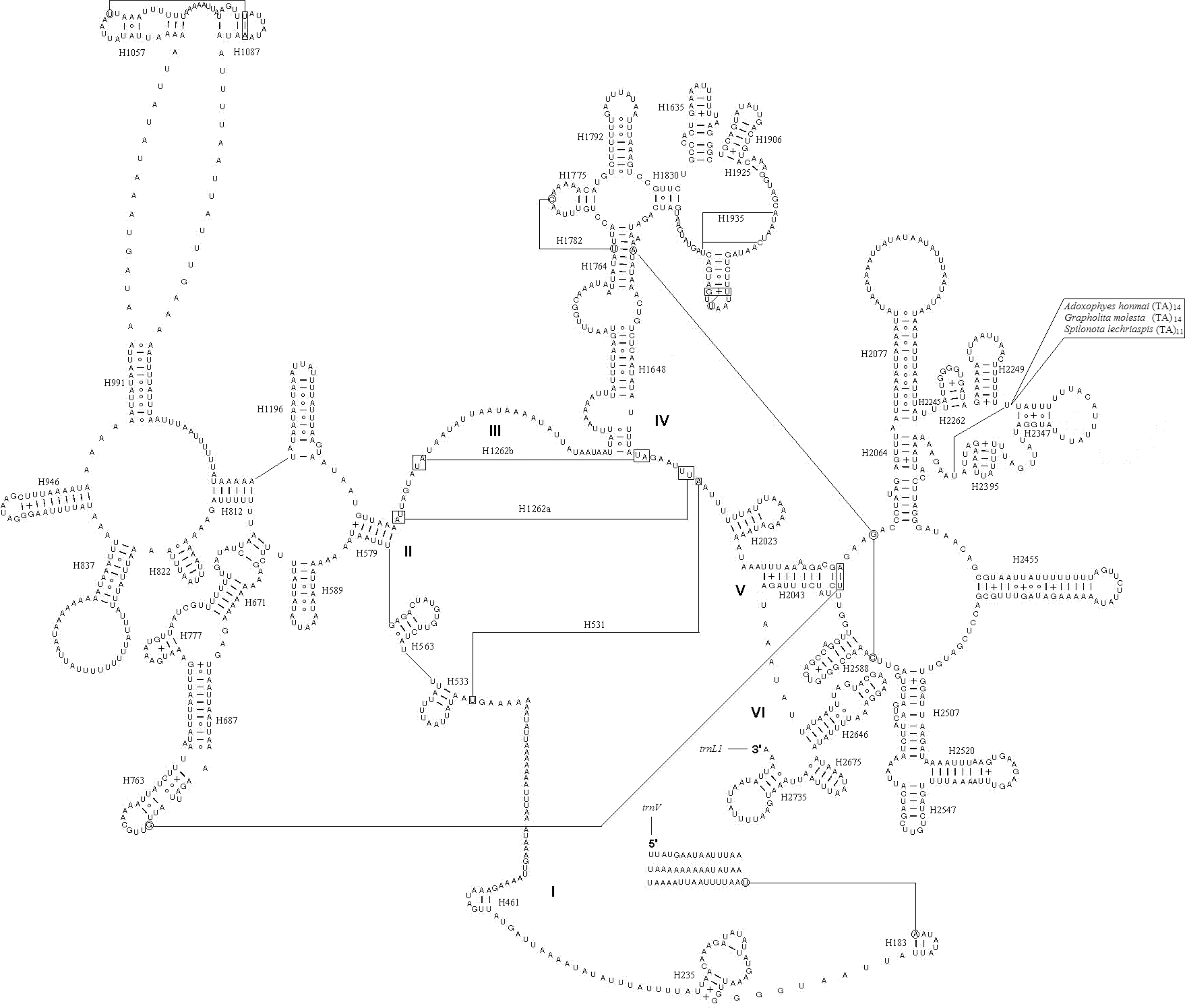
Predicted rrnL secondary structure in Leucoptera malifoliella mitogenome.

Predicted rrnS secondary structure in Leucoptera malifoliella mitogenome. Tertiary interactions and base triples are shown connected by continuous lines. Base pairing is indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by plus, and other noncanonical pairs by circles.
Codon usage
Relative synonymous codon usage values of the L. malifoliella mitogenome are summarized in Table 4. The codons CUG, ACG, and GCC were not represented in the coding sequences. The most frequent amino acids in L. malifoliella mitochondrial proteins are leucine (14.4%), isoleucine (12.8%), phenylalanine (10.9%), and serine (8.6%).
A total of 3721 codons were analyzed, excluding the initiation and termination codons.
The amino acids encoded by codons are labeled according to the IUPAC-IUB single-letter amino acid codes.
RSCU, relative synonymous codon usage.
Noncoding and overlapping region
The L. malifoliella mitogenome harbors 16 noncoding regions, ranging from 1 to 43 bp. Intergenic spacer sequences have 5 regions with a length of more than 14 bp. The remaining intergenic spacers were less than 11 bp.
Spacer 1 (43 bp) is located between the trnQ and nad2 genes. This spacer can be taken as lepidopteran feature, not found in other insects. Kim et al. (2009) detected high sequence identity between the intergenic spacer sequence and the neighboring nad2 from several lepidopteran insects; this indicated that the spacer sequence may have originated from a partial duplication of the nad2 gene.
Spacer 2 (19 bp) is found between nad3 and trnA gene; the spacer is longest in lepidopterans sequenced, and in others it is only 1 to 2 bp. Additionally, this region is generally overlapped in other lepidopterans, such as B. mandarina, B. mori, M. sexta, O. furnacalis, O. nubilalis.
Spacer 3 (21 bp) is found between the nad5 and trnH genes; the spacer is also found in A. honmai (23 bp), E. pyretorum (18 bp), B. mandarina (18 bp), B. mori (21 bp), A. melete (18 bp), and C. raphaelis (16 bp). Spacer 4 (14 bp) is found between the cob and trnS2(UCN) genes. This spacer is also present in A. pernyi (15 bp), A. yamamai (24 bp), S. boisduvalii (41 bp), and M. sexta(21 bp), and it is shorter in other lepidopterans.
Spacer 5 (19 bp) is between the trnS2(UCN) and nad1 genes, commonly detectable in lepidopterans with size 16–38 bp. This intergenic spacer is conserved for all insects. Most lepidopterans harbor the motif (ATACTAA), except for ATACTAT in Corcyra cephalonica (unpublished, HQ897685) and ATCATAT in Sesamia inferens (unpublished NC_015835). Similarly, in Hymenoptera there is a 6-bp conserved motif (THACWW) (Wei et al., 2010). In Coleoptera, there is a 5-bp conserved motif (TACTA) (Sheffield et al., 2008). The motif has been suggested to be a possible mitochondrial transcription termination peptide-binding site (Taanman, 1999).
Overlapping sequences had a total length of 25 bp from 1 to 8 bp, spread over 14 regions. The longest overlapping sequence AAGCCTTA (8 bp) is located between the trnW gene and the trnC gene. The seven-nucleotide overlap (ATGATAA) is located between atp8 and atp6, which is common in other insects. The remaining overlapping sequences are less than 3 bp.
A+T-rich region
The A+T-rich region of L. malifoliella mitogenome is located between rrnS and trnM, with 95.36% AT nucleotides and a length of 733 bp. There is a motif ATAGA downstream of rrnS, but not followed by the typical poly (T) stretch, but replaced by a stem-loop structure (Fig. 5). There are three long repeat (154 bp) sequences followed by one short repeat (56 bp), each preceded by (TA)n microsatellite regions (Fig. 5). Finally, a 10-bp poly-A is present upstream of trnM, a feature common across lepidopterans.
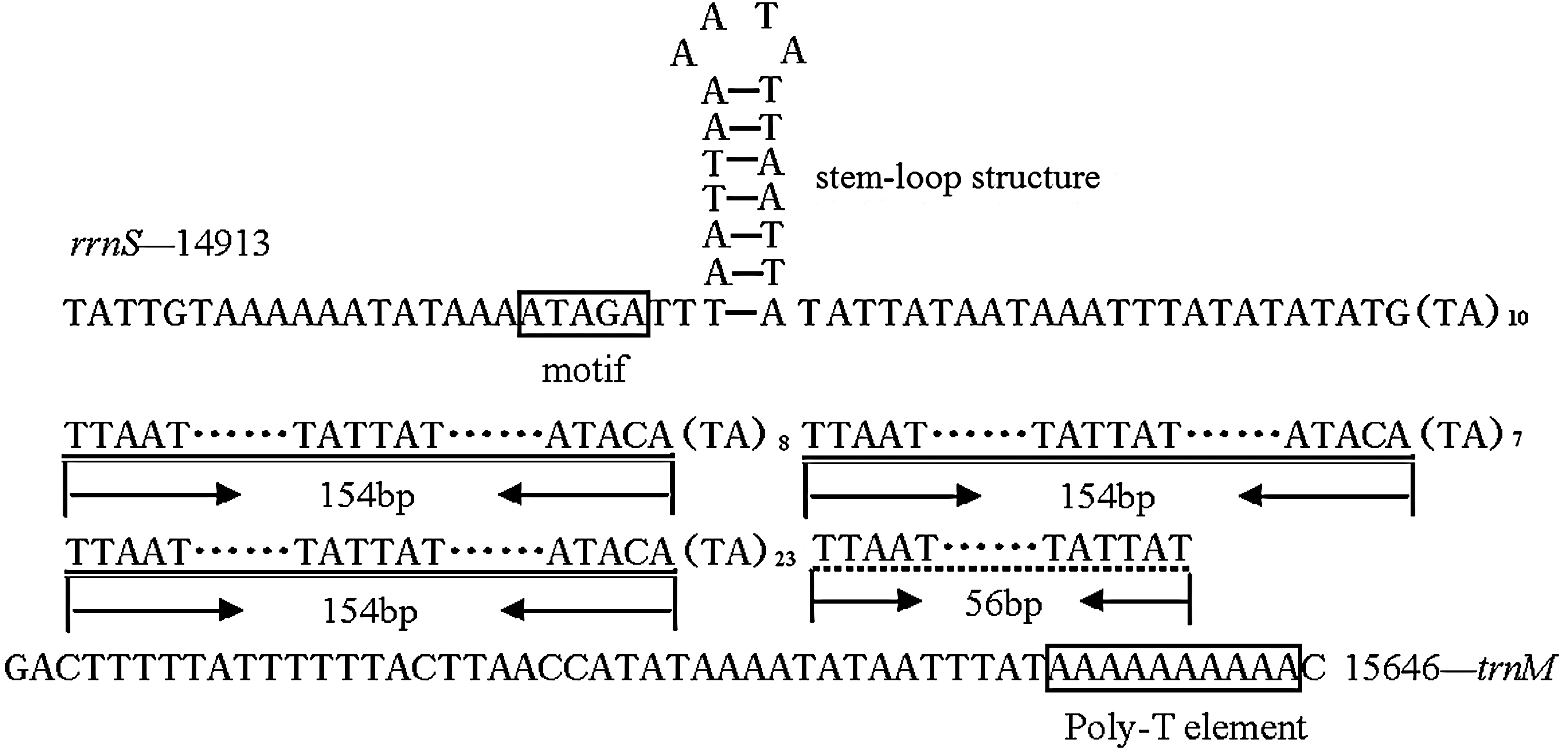
The structure of the A+T-rich region of Leucoptera malifoliella mitogenome.
The stem-loop structure in the A+T-rich region was also observed in other insect orders, including Orthoptera, Diptera, Plecoptera, Hymenoptera, and Phthiraptera (Brehm et al., 2001; Schultheis et al., 2002; Cameron et al., 2007; Cha et al., 2007; Ye et al., 2008). The stem-loop structure in the A+T-rich region of Drosophila was suggested as the site of the initiation of light strand synthesis (Clary and Wolstenholme, 1987), but the position of the stem-loop structure in L. malifoliella is found to be same as the poly (T) stretch of other lepidopterans (Fig. 6), a feature only found in Leucoptera. Two species (A. yamamai and S. boisduvalii) in Lepidoptera have a stem-loop structure, but also possess a poly (T) stretch, and the flanking sequence of the stem-loop structure are conserved, with consensus TATA sequences at the 5′ and G(A)nT at the 3′. The feather is also among other insects (Zhang et al., 1995; Schultheis et al., 2002). In contrast to these insects, there are no conserved sequences flanking both sides of the L. malifoliella stem-loop structure, and the location of stem-loop structure is closer to rrnS. Ye et al. (2008) suggested that the stem-loop structure might have the same function as the poly (T) stretch, if the latter feature is absent. Therefore, the stem-loop structure in L. malifoliella may play an important role in recognition of the light strand replication origin, but determining the function needs additional research.

Alignment of motif and Poly(T) in the A+T-rich region of 34 lepidopterans. The Poly(T) stretch is marked gray. Marked box is motif ATAGA. Underline is the stem-loop structure of Leucoptera malifoliella.
Phylogenetic Relationships
To place the L. malifoliella mitogenome relative to other lepidopterans mitogenomes and investigate the phylogenetic relationships among the superfamilies in Lepidoptera, two data sets containing the concatenated amino acid sequences and nucleotide sequences of 13 PCGs were generated. These 34 sequences represent seven superfamilies: Bombycoidea, Geometroidea, Noctuoidea, Papilionoidea, Pyraloidea, Tortricoidea, and Yponomeutoidea. According to the most recent consensus view of lepidopteran relationships in Kristensen and Skalski (1999), Papilionoidea, Bombycoidea, Noctuoidea, and Geometroidea are designated as the Macrolepidoptera; Pyraloidea together with Macrolepidoptera are designated as Obtectomera; Tortricoidea together with Obtectomera are designated as Apoditrysia; Yponomeutoidea is the sister to the remaining lepidopteran superfamilies covered in the present study. The BI and ML analyses generate similar topologies, and most major groups were consistently monophyletic apart from Pyraloidea. Three trees all support that C. cephalonica is grouped with Pyraloidea; this is same as traditional classifications (Solis, 1997). However, in the BI tree inferred from amino acid sequences, C. cephalonica is sister to the clade (Pyraloidea+(Noctuoidea+(Geometroidea+Bombycoidea))).
In our phylogenetic results, the placement of Yponomeutoidea (as represented by L. malifoliella) is the same as the traditional classification, basal to all Lepidoptera, with full nodal support in BI (100%/100%) and ML analyses (100%/100%). Bombycoidea and Geometroidea are sister groups with high nodal support on BI (100%/100%) and ML analyses (90%/92%) (Fig. 7A, B), which is consistent with Yang et al. (2009), but differs to the typical morphological results, which give a sister group relationship between the Papilionoidea and Geometroidea. Papilionoidea is the sister of the remaining macrolepidopteran families, in accordance with other studies (Jiang et al., 2009; Yang et al., 2009; Liao et al., 2010). Pyraloidea has a closer relationship to most Macrolepidoptera than Papilionoidea (butterflies), a result confirmed by a recent study (Regier et al., 2009), but different from the traditional classification.

Phylogeny of lepidopteran insects.

Footnotes
Acknowledgments
Prof. Qi-lian Qin and his lab members (Institute of Zoology, Chinese Academy of Sciences) kindly provided advice and facilities in sequence cloning. We also thank Shu-jun Wei (Institute of Plant and Environmental Protection, Beijing Academy of Agriculture and Forestry Sciences) and Xiao-he Wang (Institute of Zoology, Chinese Academy of Sciences) for their kind help in data analysis.
This work was supported mainly by grants from the Knowledge Innovation Program of Chinese Academy of Sciences (Grant No. KSXC2-EW-B-02), Public Welfare Project from the Ministry of Agriculture, China (Grant No. 201103024), the National Science Foundation, China (NSFC Grant No. 30870268, 31172048, J0930004) to Chao-dong Zhu and NSFC Grant (No. 31172129) to Chun-Sheng Wu.
Disclosure Statement
No competing financial interests exist.