
Abstract
Cancer is a collection of very complex diseases that share many traits while differing in many ways as well. This makes a universal cure difficult to attain, and it highlights the importance of understanding each type of cancer at a molecular level. Although many strides have been made in identifying the genetic causes for some cancers, we now understand that simple changes in the primary DNA sequence cannot explain the many steps that are necessary to turn a normal cell into a rouge cancer cell. In recent years, some research has shifted to focusing on detailing epigenetic contributions to the development and progression of cancer. These changes occur apart from primary genomic sequences and include DNA methylation, histone modifications, and miRNA expression. Since these epigenetic modifications are reversible, drugs targeting epigenetic changes are becoming more common in clinical settings. Daily discoveries elucidating these complex epigenetic processes are leading to advances in the field of cancer research. These advances, however, come at a rapid and often overwhelming pace. This review specifically summarizes the main epigenetic mechanisms currently documented in solid tumors common in the United States and Europe.
Common Themes in Cancer Epigenetics
DNA methylation
DNA methylation in humans is carried out by three enzymes: DNA methyltransferase 1 (DNMT1), which maintains parental methylation patterns, and DNMT3A and DNMT3B, which regulate de novo methylation (Bestor and Ingram, 1983; Okano et al., 1999; Chen et al., 2007; Chedin, 2011). As detailed next, these enzymes are differentially expressed in certain cancers, but their expression does not always correlate with hyper- or hypomethylation, as one would expect. This may be partially explained by the fact that DNMT expression is sometimes regulated by miRNAs (described next, Fig. 1).
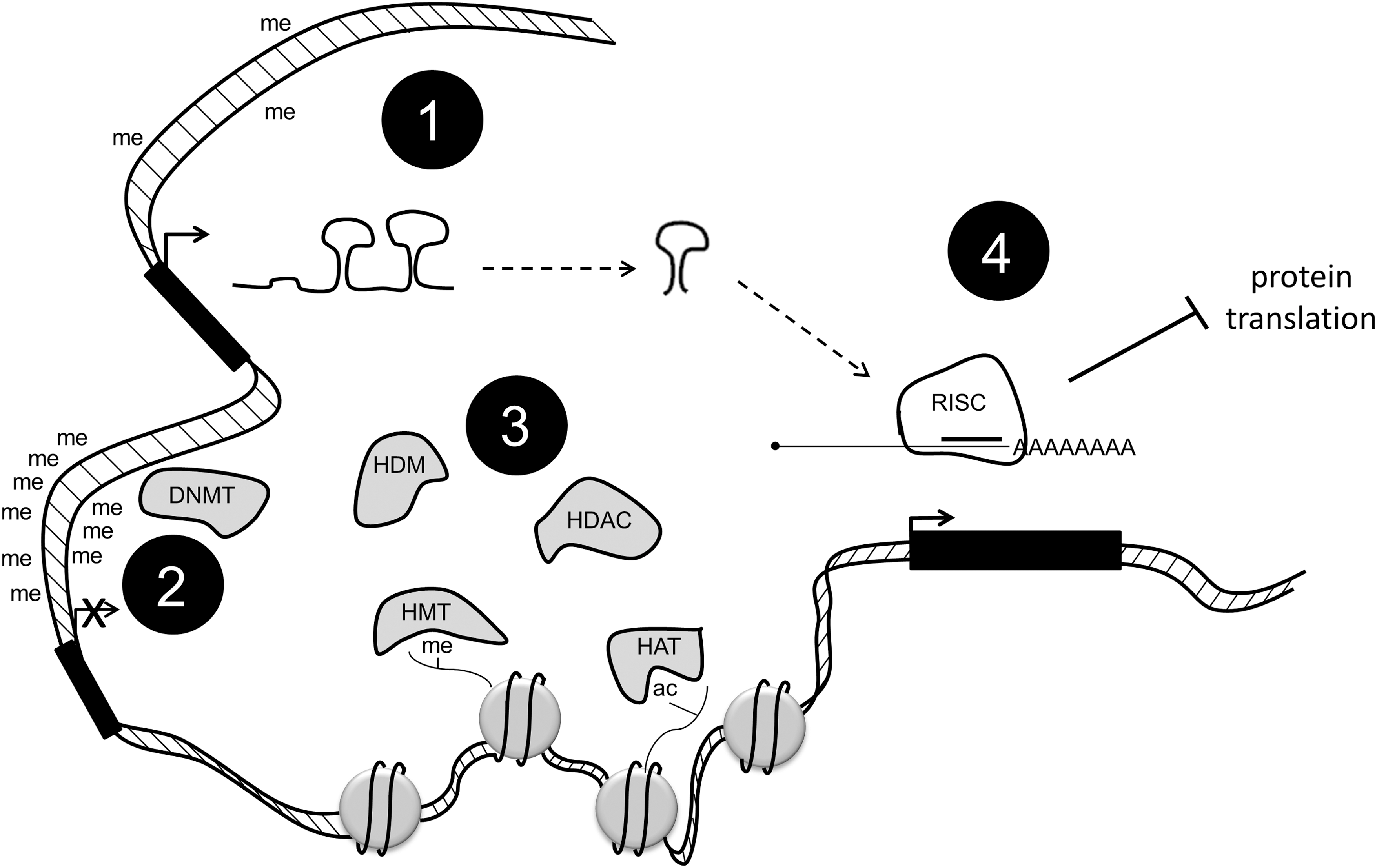
DNA methylation, histone modification, and micro RNA (miRNA) expression work together to control the complex environment of epigenetics in cancer. Promoter hypomethylation is linked to the expression of genes, including miRNA, DNA methyltransferases (DNMTs), and histone modifiers (1). DNMTs can, in turn, hypermethylate promoters and turn off these same genes (2). Histone-modifying enzymes can affect gene expression by adding or removing certain marks (3). miRNAs are processed and targeted to mRNA, leading to a decrease in the protein expression of certain enzymes (4).
Active loss of DNA methylation has been observed during development, suggesting the existence of DNA demethylases. Multiple mechanisms for DNA demethylation have been proposed and recently discussed (Wu and Zhang, 2010). In particular, one potential mechanism is through Ten-eleven translocation 1–3 (TET1-3) proteins, which are a group of hydroxylases (Tahiliani et al., 2009; Ito et al., 2011). These enzymes can convert 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (Ito et al., 2010; He et al., 2011; Pfaffeneder et al., 2011). These modified bases may not only serve as intermediates in the DNA demethylation process, but they may also increase the diversity of the epigenetic states of genomic DNA. The TET1 protein was identified as a fusion partner of mixed lineage leukemia (MLL) in patients with acute myeloid leukemia (AML), while mutations of TET2 proteins have been found in patients with various myeloid malignancies (Wu and Zhang, 2011).
Histone modification
Another way that cancers are epigenetically regulated is through alterations in histone modification. In each cell, nucleus DNA is wrapped around an octamer of histone proteins. These proteins have flexible N-terminal tails that are extensively modified (Fig. 1). Certain modifications serve to tighten or loosen DNA from the octamer, thus allowing or disallowing the binding of transcriptional machinery. This enables a tight control of gene expression. Some histone modifications are associated with active genes (i.e., trimethylation on histone H3, lysine 4 [H3K4me3]), and some are associated with repressed genes (i.e., H3K9me3) (reviewed in Fullgrabe et al., 2011). The enzymes responsible for adding and removing these marks are differentially expressed in many cancers (Chi et al., 2010; Blair et al., 2011).
The most common histone modifications are acetylation, methylation, and phosphorylation. Acetyl groups are added to lysine residues by enzymes called histone acetyltransferases (HATs) and are removed by histone deacetylases (HDACs). Methyl groups are added to lysine and arginine residues by histone methyltransferases (HMTs) and are removed by histone demethylases (HDMs). The roles of these histone-modifying enzymes in cancer are still not completely understood, but some have been linked to specific diseases, as discussed next.
Micro RNAs
Recent discoveries have highlighted the importance of microRNAs (miRs) in the regulation of cancer (Braconi et al., 2011; Fanini et al., 2011; Ferracin et al., 2011; Landau and Slack, 2011). miRs are small noncoding RNAs that are highly processed using specific machinery (Fig. 1). Once processed down to mature miRNAs, they are targeted to 3′UTRs of mRNA, where they inhibit the translation of the targeted mRNAs and/or cause those mRNAs to be targeted for degradation. This leads to a decrease in the amount of protein made. Either miRs can be oncogenic or they can have tumor suppressive functions. More recently, a push for diagnostic and prognostic miRs has been established due to the identification of many differentially expressed miRs in cancers as compared with normal tissue (Kasinski and Slack, 2011).
Epigenetic crosstalk
Interestingly, all three types of epigenetic regulation just mentioned are somewhat intertwined (Fig. 1). DNA demethylases are post-transcriptionally regulated by miR-29 and miR-148 (Fabbri et al., 2007; Duursma et al., 2008). DNMTs have also been shown to interact with HDACs (Yang et al., 2001; Bai et al., 2005). In fact, DNMT and HDAC inhibitor combinations are at the forefront of cancer therapies (Zhu and Otterson, 2003; Unoki, 2011). Additionally, miR expression is regulated by DNMT-mediated promoter methylation (So et al., 2011).
Epigenetic markers show promise as biomarkers for many different reasons. Isolating genomic DNA for methylation profiling is easier, and the DNA is more stable than extracting mRNA for gene expression analysis. Strides have been made in profiling clinical samples for histone marks using immunohistochemistry and mass spectrometry. Studying the enzymes that alter DNA methylation and histone modification patterns may be the key to curbing cancer progression. Next, we summarize the advances in the epigenetic mechanisms of DNA methylation, histone modifications, and miRNA expression in six different cancers: breast, prostate, melanoma, pancreatic, lung, and colorectal.
Breast Cancer
Women in the United States have a one in eight chance of being diagnosed with some type of breast cancer in their lifetime. The disease has very personal connotations to many people and, as a result, greater efforts have been dedicated to investigating breast cancer than many other cancers. This has led to a plethora of literature in recent years on the mechanisms of breast cancer. Many of the concepts and mechanisms discovered in breast cancer are being applied to the research of other cancers. Much progress has been made in the education, early detection, and treatment of breast cancer, but a few drugs have been developed to treat specific forms of the disease. One such drug is Herceptin/trastuzumab, which targets human epidermal growth factor receptor 2 (HER2/ERBB2), a receptor tyrosine kinase (Vogel et al., 2002). Many patients have responded to this medication, and it has undoubtedly saved lives. However, there are many different forms of breast cancer, and the development of drugs that are specific to mechanisms within these subtypes will allow for a type of personalized medicine with higher specificity and less severe side effects. Although epigenetics is a relatively young field, much has been published about breast cancer epigenetics.
There are currently six major sub-types of breast cancer and within these sub-types, individual cases differ (Chin et al., 2006; Prat et al., 2010; Stecklein et al., 2012). Breast cancers are often classified by their estrogen receptor (ER/ER-α/ESR1), progesterone receptor (PR), and HER2 status. Patients negative for all three are classified as triple negative, which is a very aggressive and devastating form that is difficult to treat. ER positive tumors often respond to hormone therapy, and HER2+ tumors have shown some success with Herceptin. It is crucial to attain a better understanding of the different types of breast cancer in order to provide patients with the best care possible.
Similar to many tumors, breast cancers have been shown to be globally hypomethylated (Soares et al., 1999). On a more detailed study of CpG islands, however, it becomes clear that many tumor suppressor genes are hypermethylated in cancer compared with their noncancerous counterparts (Hinshelwood and Clark, 2008). The suppression of tumor suppressor genes is a common mechanism that is used in the progression of cancer. DNA hypermethylation is generally associated with gene instability due to the downregulation of tumor suppressors in DNA repair pathways in breast cancer (Widschwendter and Jones, 2002). Some gene-specific methylation may be accounted for by the fact that DNMTs are overexpressed in breast cancer, specifically DNMT3B, but this remains a loose correlation (Girault et al., 2003).
Some gene methylation changes have been linked to specific types of breast cancer. For example, ER promoter methylation is linked to a loss of ER expression (Weigel and deConinck, 1993; Ferguson et al., 1995). Breast cancers overexpressing ER and PR are often treated using hormone therapy involving drugs that mimic estrogen or block these receptors. Methylation patterns have also been used more recently to determine a patient response to a certain treatment. For example, patients using Tamoxifen are profiled for the methylation levels of circulating Ras-association domain family 1 isoform A (RASSF1A). If the levels of RASSF1A methylation are elevated, then it indicates that the patient is not responding to the treatment (Fiegl et al., 2005). This is a valuable tool in determining how to proceed with individual patients.
Since DNA hypermethylation is partially driven by the overexpression of DNMTs, they have also become targets for cancer drugs. Traditionally, DNMT inhibitors are nucleoside analogs that are incorporated into DNA and serve to permanently link the DNMTs to the DNA. These DNMT/DNA hybrid molecules are then targeted to the proteasome for degradation. Some of these inhibitors have been approved for clinical use, although none specifically for breast cancer (Foulks et al., 2011).
The role of histone-modifying enzymes in breast cancer has become more apparent in recent years (reviewed in Huang et al., 2011). The majority of breast cancer treatment advances involving histone-modifying enzymes have come in the form of HDAC inhibitors. In general, histone acetylation is linked to the activation of genes. It is thought that acetylation causes loosening of the DNA around the histones, which allows transcriptional machinery to access the DNA. The removal of acetylation, conversely, is thought to cause the DNA to tighten around the histones and to inhibit the access of transcription machinery and silencing genes. HDACs have been shown to remove these activating marks from tumor suppressor genes, thereby reducing their expression and leading to the development of cancer. HDAC inhibitors have shown some clinical promise in breast cancer patients. For example, Vorinostat has entered phase II clinical trials for metastatic breast cancer in combination with other anticancer drugs (Ramaswamy et al., 2011).
More recently, emphasis has also been placed on determining the role of HMTs and HDMs in cancer. An alteration in the expression and regulation of these genes has been noted in breast cancers. Enhancer of zeste homolog 2 (EZH2) is an H3K27 methyltransferase that has been shown to be up-regulated in breast cancers (Kleer et al., 2003). Lysine-specific demethylase 1 (LSD1) is an H3K4 demethylase that is highly expressed in ER negative tumors and has been suggested as a potential predictive marker for aggressive breast cancers (Lim et al., 2010). LSD1 also interacts with HDACs, which suggests that a combination of demethylase inhibitors and HDAC inhibitors could be a potent cancer treatment (Huang et al., 2011). Interestingly, LSD1 has been shown to regulate the methylation of both p53 and DNMT1, which could suggest a key role in other cancer mechanisms (Huang et al., 2007; Wang et al., 2009).
Since the discovery of LSD1 in 2004, more HDMs have been identified, and many of them appear to play instrumental roles in breast cancer (Blair et al., 2011). For example, lysine demethylase 5B (KDM5B, also known as PLU1 and JARID1B), an H3K4me3 demethylase that is overexpressed in breast cancer, has been shown to repress tumor suppressor genes such as BRCA1 and CAV1 in breast cancer cell lines (Yamane et al., 2007). The Taylor–Papadimitriou lab showed a direct interaction between HDAC4 and KDM5B, which they suggest to be a potential mechanism for KDM5B in cancer (Barrett et al., 2007). KDM5B has also been implicated in mammary development and is becoming a major player in breast cancer research (Catchpole et al., 2011).
There are currently estimated to be many differentially expressed miRNAs in breast cancer, many of which are noted in Table 3 (Veeck and Esteller, 2010). Let-7 is down-regulated in breast cancers, and its overexpression in these cancers leads to a decrease in metastatic potential and proliferation rate, suggesting that Let-7 is a key player in the disease (Yu et al., 2007). miR-125 targets HER2 and is, therefore, thought to be a tumor suppressor in HER2+ breast cancers (Scott et al., 2007). Many miRNAs have been suggested to be potential diagnostic or prognostic markers for breast cancer; for example, circulating levels of miR-195 have been noted to be up-regulated in breast cancer patients (Heneghan et al., 2010; Ferracin et al., 2011). Recent studies by Ma et al. (2010) outline the potential for miR-targeted therapies, so called “antago-miRs,” in treating the disease. They show that antagomiR-10b aids the prevention of lung metastases in a mouse breast cancer model (Ma et al., 2010). Additionally, many miRs have been directly associated with the different stages of metastasis and are, therefore, of interest in all types of cancers, specifically those that are prone to metastasis (reviewed in (Shi et al., 2010).
miRNA, micro RNA.
Prostate Cancer
Prostate cancer is one of the most commonly occurring cancers in men in the United States and Europe (Jemal et al., 2010; Siegel et al., 2011). In the absence of metastasis, the disease is often slow progressing and can sometimes be treated by a “watch and wait” procedure. Since this type of prostate cancer is usually androgen regulated, many treatments similar to breast cancer, such as hormone deprivation, are used to control the disease (Shepard and Raghavan, 2010; Corona et al., 2011). However, there is still no effective treatment method against metastatic prostate cancer, and better ways to diagnose and treat the disease are crucial to curing it. Although most cancers have been shown to be globally hypomethylated, prostate cancer is generally an exception to this rule (Jeronimo et al., 2011). More than 50 genes are hypermethylated in the disease, some of which are highlighted in Table 2. Many of these genes are involved in important cellular pathways and present relatively early in the disease stage. Additionally, urokinase plasminogen activator (PLAU) is highly hypomethylated in castration-resistant prostate cancers (CRPCs) (Pakneshan et al., 2003). A thorough understanding of which genes are differentially methylated in which types of prostate cancer could lead to early diagnosis and improve treatment outcomes (Van Neste et al., 2011). Studies focusing on detailing the differential methylation patterns of these diseases are ongoing, and information about the mechanisms behind these aberrant methylation patterns is emerging (reviewed in Park, 2010). Some of these studies outline the effect of certain factors, including age, diet, and environment, on promoter methylation (Venkateswaran and Klotz, 2010). While changing a person's ethnicity or age is not an option, information linking diet and prostate cancer epigenetics has great potential for preventative purposes.
Histone-modifying enzymes have also been implicated as being important in prostate cancer. Similar to breast cancer, the most commonly studied histone modifying enzymes in prostate cancer are EZH2, HDACs, and LSD1. EZH2 overexpression leads to promoter hypermethylation in prostate cancer, which results in the repression of certain developmental and tumor suppressor genes (Hoffmann et al., 2007). HDACs are overexpressed in prostate cancer, particularly so in CRPC (Halkidou et al., 2004). In addition, androgen receptor (AR) in CRPC has recently been shown to mediate its own expression by recruiting LSD1 to the AR gene and suppressing expression through H3K4 demethylation (Cai et al., 2011).
The profiling of prostate cancer cells has led to the discovery of 50 differentially regulated miRs (reviewed in Catto et al., 2011). Only a few of these targets have been extensively studied, such as miR-221/222. The overexpression of miR-221 or miR-222 in LNCaP prostate cancer cells leads to a reduction in prostate-specific antigen (PSA), which is currently the most common biomarker for prostate cancer (Sun et al., 2009). Although testing PSA levels in serum has been useful in diagnosis, the test is not specific and leads to many false positives and false negatives (Croswell et al., 2011). This has led to the push for a better biomarker for prostate cancer screening. As in other cancers, miRNAs are exciting potential targets for such a test. Specifically, miR-200a has been shown to be differentially expressed in men who relapse from radical prostatectomy (Barron et al., 2011). Transient transfection of miR-200a significantly reduces the proliferation of prostate cancer cells, suggesting that it might be a potential target for therapies (Barron et al., 2011).
Apart from serving as prognostic and diagnostic markers, epigenetic marks in prostate cancer also show promise as therapeutic targets. Clinical trials using DNMT inhibitors in prostate cancer have shown minimal benefit, but these trials were performed in patients with advanced stage, hormone independent disease (Thibault et al., 1998). More trials are needed using these inhibitors alone and in combination with other epigenetic inhibitors. HDAC inhibitors have been used in clinical trials for patients with advanced-stage prostate cancer (Munster et al., 2009). Their success has been highest in trials that combine them with conventional chemotherapeutic drugs. HMT and demethylase inhibitors have shown promise in in vitro prostate cancer systems and are currently under development for use in a clinical setting (Tan et al., 2007; Miranda et al., 2009).
Melanoma
Many skin cancers are benign and, if caught early enough, can be removed in a simple procedure, often performed in the doctor's office. In recent years, skin cancer prevention and screening have become more popular, and the public is becoming more aware of the dangers of UV exposure and other causes of the disease. The incidence of melanoma, however, has risen in recent years (Linos et al., 2009). Melanoma is considered the most devastating form of skin cancer due to the high mortality associated with the disease. Early detection is key, because once this type of cancer metastasizes, then a patient's life expectancy is around 2 years (Koon and Atkins, 2007). Cases of melanoma are on the rise around the world, and it has become more important to understand the molecular mechanisms of this disease in order to improve early detection methods and treatments.
Interestingly, hypomethylation has been discovered not only globally but also in certain genes in melanoma (Table 1). These genes tend to be oncogenic in nature; therefore, hypomethylation of their promoters causes overexpression and leads to cancer progression. A recent study outlines the importance of mutant BRAFV600E in hypomethylation and, thus, the activation of the genes associated with cell proliferation and invasion in melanoma cell lines (Hou et al., 2012).
More than 50 genes have been reported as being hypermethylated in melanoma (Table 2). The function of some of these genes remains unknown, but many are associated with processes involved in cancer progression such as apoptosis and cell-cycle regulation (Rothhammer and Bosserhoff, 2007). Promising results point to the use of DNA methylation levels as melanoma biomarkers. For example, circulating levels of ER methylation can be associated with poor prognosis (Mori et al., 2006). Additionally, a methylation signature could be established for diagnosing melanoma. Reports show that suppressor of cytokine signaling 1 and 2 (SOCS1 and SOCS2), retinoic acid receptor β-2 (RARβ2), and tumor necrosis factor receptor superfamily, members 10c and 10d (TNFRSF10C and TNFRSF10D) are frequently methylated in clinical melanoma samples, although further validation of these methylation patterns is needed (Howell et al., 2009). DNMTs have also been targeted in melanoma patients. Studies have shown that DNMT inhibitors can slow the progression of melanoma in nude mouse models (Morita et al., 2006).
Histone-modifying enzymes have also been studied as being possible targets for melanoma treatment. Melanoma tumor suppressor genes inhibitor of growth 3 and 4 (ING3 and ING4) have been linked to lysine acetyltransferase 7 and 8 (KAT7 and KAT8), which are components of a larger histone acetyltransferase complex (Li et al., 2008). In addition, the HAT components p300 and CPB have been linked to senescence in melanoma cells (Bandyopadhyay et al., 2002). These factors act by altering the expression of microphthalmia-associated transcription factor (MITF), a master regulator of melanocyte development and melanomagenesis (Sato et al., 1997; Price et al., 1998; Bandyopadhyay et al., 2002). Although the mechanisms of HDAC inhibitors are not well understood, it is thought that they can allow re-expression of tumor suppressor genes in melanoma and increase the acetylation levels of some nonhistone targets (Howell et al., 2009). One particular HDAC inhibitor, MS-275, has shown promise in clinical trials with melanoma patients (Gore et al., 2008).
Some more recent reports have outlined the importance of KDM5B in melanoma. A population of slow-cycling and stem-like melanoma cell lines expressed high levels of KDM5B, indicating that it is involved in making melanoma cells more “stem-like” (Roesch et al., 2010). The exact mechanism by which KDM5B contributes to the disease is still under investigation, and drugs targeting the enzyme are currently under development.
Several miRNAs have been implicated in melanoma progression. miR-137, for example, is expressed in melanoma cell lines and targets MITF (Levy et al., 2006). MITF acts by regulating the differentiation and cell-cycle genes, and the binding of miR-137 prevents these mRNAs from being translated (Bonazzi et al., 2011). Let-7 is down-regulated in melanomas, and studies show that loss of let-7a is linked to the development and progression of melanoma (Muller and Bosserhoff, 2008; Schultz et al., 2008). miR-221 and miR-222 are regulated by the transcription factor promyelocytic leukemia zinc finger (PLZF) in melanoma. By binding the regulatory regions of miR-221/222, PLZF inhibits their ability to target mRNA, such as c-Kit Receptor and p27/CDK5N1B. By inhibiting the degradation of these tumor suppressors, PLZF suppresses melanoma progression (Felicetti et al., 2008).
Pancreatic Cancer
Due to the lack of symptoms and difficulty in attaining early diagnoses, pancreatic cancer is one of the deadliest forms of cancer. At the time of diagnosis, most patients have an unresectable form of the disease and have a less than 2% 5-year rate of survival (Jemal et al., 2010; Siegel et al., 2011). Although progress has been made in recent years, better ways to detect and treat pancreatic cancer are needed. The only current prevention measures suggested are those recommended for all cancers: smoking cessation, exercise, and a diet that is high in fruits and vegetables. Some cases have been linked to hereditary causes, but the specific genes that are involved have yet to be identified (Matsubayashi et al., 2011).
Due to the correlation between late diagnosis and increased mortality, finding a biomarker associated with the disease is crucial. Therefore, determining the epigenetic mechanisms of the disease is useful. Investigations into DNA methylation have shown that 80% of pancreatic cancers overexpress DNMT1 (Peng et al., 2005). Some tumor suppressor genes such as CDKN2A/p16, MutL homolog 1 (MLH1), and cadherin 1 (CDH1) are hypermethylated in pancreatic cancer (Schutte et al., 1997; Ueki et al., 2000). Proenkephalin (PENK) is aberrantly methylated in 90% of pancreatic cancers and reprimo (RPRM), in 80% (Fukushima et al., 2002; Sato et al., 2006). Tumor suppressor CDKN2A is often used as an example of epigenetics in pancreatic cancer (Lomberk, 2011). It is hypermethylated and, therefore, silenced in pancreatic cancer (Singh and Maitra, 2007). Many genes silenced in pancreatic cancer are also hypermethylated in precursor lesions, indicating that the screening for DNA methylation patterns could be a reliable basis for early diagnosis (Sato et al., 2005). As seen in Table 1, some genes have been shown to be hypomethylated in pancreatic cancer, but the mechanism behind this phenomenon is not well understood (Sato et al., 2003).
Recent studies have outlined the importance of chromatin-associated proteins in pancreatic cancer. The polycomb protein BMI1 is recruited to CDKN2A, where it stalls EZH2, allowing for the hypermethylation of H3K27 at the CDKN2A promoter, leading to the repression of CDKN2A (Bracken et al., 2007). Additionally, Jiao et al. (2011) used exome sequencing to discover common mutations in alpha thalassemia/mental retardation syndrome X-linked (ATRX) and death-domain-associated protein (DAXX) in pancreatic neuroendocrine cancers (panNETs). Both ATRX and DAXX are involved in chromatin remodeling (Jiao et al., 2011). They work together to deposit histone variant H3.3 at regions of silent chromatin (Goldberg et al., 2010). They also discovered mutations in menin (MEN1), which regulates histone methylation by recruiting the HMT MLL (Jiao et al., 2011).
More than 50 miRNAs have been associated with pancreatic cancer (reviewed in Wang and Sen, 2011). Some of these miRs, such as miR-155, miR-221, and miR-222, are also overexpressed in other cancers. However, some unique miRNAs, such as mIR-376a and miR-301, have been discovered as being expressed in pancreatic cancer (Lee et al., 2007). Although follow-up literature on pancreatic cancer is not currently available, other reports show that miR-376a is associated with erythroid differentiation, and miR-301 can promote invasion in breast cancer (Shi et al., 2011; Wang et al., 2011). A recent report by Wang et al. (2011) uses 11 miRNAs deregulated in pancreatic ductal adenocarcinoma (PDAC) to determine the most common cellular pathways that are affected by these changes (Wang and Sen, 2011). They showed that major components of the phosphatase and tensin homolog (PTEN) signaling pathway are affected by the down-regulation of these miRs. PTEN is a tumor suppressor whose function is lost in many cancers, including pancreatic cancer (Krausch et al., 2011). Some miRNA expression profiles, specifically the down-regulation of miR-217 and the overexpression of miR-196a, have even been suggested for their use to diagnose pancreatitis versus PDAC (Bloomston et al., 2007; Lee et al., 2007; Szafranska et al., 2007).
Lung Cancer
The leading cause of cancer deaths worldwide is lung cancer. The outcome for patients with lung cancer is not a good one. The 5-year survival rate is around 13%. Unfortunately, a stigma is attached to having the disease. Many lung cancer patients have smoked for much of their lives, and many continue to even after treatment; however, many lung cancer patents are never-smokers. This stigma has led to a lack of sympathy for these patients as compared with patients with other types of cancer and has translated to a difference in the level of funding for lung cancer research compared with other common cancers.
There are two types of lung cancer: small cell (SCLC) and nonsmall cell (NSCLC). The more common of the two is NSCLC, yet the more aggressive is SCLC. Since lung cancer is cell-type specific and it is often difficult to isolate enough cells for analysis, much epigenetic research is done using cell lines or the entire lung. For this reason, progress in lung cancer epigenetics is not as extensive as in some other cancers.
Environmental factors have a more pronounced contribution to lung cancers than to most other cancers due to the direct interaction of the lungs with the outside environment through respiration. The effect of environmental factors on epigenetics is well established (Bell and Beck, 2010; Kabesch et al., 2010; Galea et al., 2011; Herceg and Paliwal, 2011; Herceg and Vaissiere, 2011; Yang and Schwartz, 2011). Some high-throughput studies of DNA methylation patterns in lung cancer have been recently reported (Ocak et al., 2009; Heller et al., 2010; Toyooka and Gazdar, 2011). A number of promoters were found to be aberrantly methylated, and validation studies are underway to verify these findings in different types of lung cancers. These validation studies aim at detailing the involvement of these aberrant methylation patterns in different types and stages of lung cancer in hopes of finding a diagnostic or prognostic marker or a therapeutic target. As in pancreatic cancer, methylation tends to occur early in lung cancer development. One interesting finding was that CDKN2A and O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation could be used to predict cancer development 3 years before clinical diagnosis (Palmisano et al., 2000). The methylation data collected to date is also being used to develop methylation profiles for diagnosis (Rauch et al., 2011).
Histone-modifying enzymes have also been shown to contribute to different types of lung cancer. Histone hypoacetylation has been recorded in NSCLC and has been linked to a differential expression of HDACs (Osada et al., 2004; Sasaki et al., 2004; Van Den Broeck et al., 2008). As a result, HDAC inhibitors have been suggested for the treatment of NSCLC (Loprevite et al., 2005; Komatsu et al., 2006; Cuneo et al., 2007; Gridelli et al., 2008). A decreased expression in HATs has also been suggested in order to explain hypoacetylation in some lung cancers (LLeonart et al., 2006; Vaquero et al., 2007). Some histone demethylase enzymes have been linked to lung cancer as well. The overexpression of KDM5B, lysine demethylase 4C (KDM4C), and JmjC-domain containing MAPJD have been noted in lung cancer and have been suggested as contributing to aberrant histone methylation levels seen in lung cancer (Italiano et al., 2006; Suzuki et al., 2007; Hayami et al., 2010). More recently, drug resistance by lung cancer cells was reported to be mediated by an increased expression of lysine demethylase 5A (KDM5A, also known as RBP2 and JARID1A) (Sharma et al., 2010).
The role of miRNAs is relatively well established in lung cancer. Some of the more commonly studied lung cancer miRs are noted in Table 3. Environmental factors such as cigarette smoke have been shown to affect miRNA expression (Hou et al., 2011; Russ and Slack, 2012). Currently, no miRNAs are used as biomarkers in the clinic. The main problem with measuring levels of miRNAs is that an accurate and sensitive method for quantitating them has not been developed (Yendamuri and Kratzke, 2011). A further study of the role of these molecules in lung cancer may lead to strides in the cancer field and is ongoing.
Colorectal Cancer
Another leading cause of cancer deaths in the United States and Europe is colorectal cancer (Jemal et al., 2010; Siegel et al., 2011). This cancer affects the large intestine of the digestive system and usually presents in polyp form. Recently, awareness and resulting heightened screening efforts have lowered the mortality rate associated with this cancer. However, it remains a potent killer once it reaches the metastatic stage. Colorectal cancers can be classified as arising from either chromosomal instability or microsatellite instability (Jass 2007; Ogino and Goel, 2008). Some treatments for the disease are available, but much more research into the molecular mechanisms of the different types of colorectal cancer is needed.
DNA methylation patterns in colorectal cancers have been studied to the extent that certain panels of methylated genes have been suggested for colorectal cancer classification (Ahlquist et al., 2008). These classifications are still under debate, but they present promising candidates for diagnostic and prognostic biomarkers. Interestingly, promoter methylation in colorectal cancers can often be associated with Kirsten rat sarcoma viral oncogene homolog (KRAS) and v-raf murine sarcoma viral oncogene homolog B1 (BRAF) mutation status. Cancers containing BRAF and KRAS mutations tend to have higher methylation levels in certain genes (Samowitz et al., 2005; Nosho et al., 2008).
While most colorectal epigenetic studies focus on DNA methylation, certain distinct histone methylation patterns have also been noted in the disease. For example, H3K9me2 levels are elevated in colorectal adenocarcinomas when compared with normal tissue and are a determinant of more aggressive forms of the disease (Nakazawa et al., 2011). Polycomb protein and HMT EZH2 is overexpressed in colorectal tumors associated with shorter survival times (Crea et al., 2011).
Some miRNAs identified as being involved in colorectal cancer include those listed in Table 3. These miRNAs function in various cellular processes that are associated with cancer, such as evasion of apoptosis, angiogenesis, epithelial-to-mesenchymal transition (EMT), differentiation, invasion, and signaling (de Krijger et al., 2011). Data on the molecular mechanisms of miRNAs in colorectal cancer are still limited, but a more extensive study of the roles of these miRNAs could lead to diagnostic and prognostic markers that could aid in the early detection or treatment.
Can We Secure a Cure?
In his recent article “Curing ‘Incurable’ Cancer,” James Watson outlines the importance of epigenetics in deciphering the mechanisms of cancer (Watson, 2011). There is a definite theme that runs through the epigenetic mechanisms of all cancers. We have outlined the link between the six cancers addressed here in Tables 4 and 5. Table 4 highlights the genes that are hypermethylated in three or more of the common cancers covered in this review. Table 5 contains the same information for miRNA expression. Studying the epigenetic links shared by common cancers provides exciting potential for a powerful anticancer drug targeting many forms of the disease. Conversely, profiling the epigenetic differences between certain cancers will allow us to design more specific drugs. Elucidating and detailing these differences and the differences in the epigenetics of diseased versus normal tissue will allow the continued development of drugs that are unique to those diseases.
Since epigenetics is a relatively young field, there is a flourish of publications daily outlining the mechanisms by which different epigenetic processes work. Some epigenetic mechanisms not mentioned here, such as chromatin remodeling and gene looping, are also being studied more extensively in the current literature and are contributing to the overall knowledge of the mechanisms of cancer not involving direct alterations in the DNA sequence. This information is currently serving and will continue to serve in the development of many different weapons in the fight against cancer. The fact that epigenetic mechanisms are generally reversible makes them excellent targets for clinical therapies. The ease of DNA isolation points to the use of DNA methylation patterns in noninvasive diagnostic and prognostic testing. The field of cancer epigenetics is one of the most logical and promising places to seek to cure the “incurable.”
Footnotes
Acknowledgments
This work was supported in part by the V Scholar Award, Breast Cancer Alliance Young Investigator Grant, Melanoma Research Foundation Career Development Award, the Alexander and Margaret Stewart Trust Fellowship, CTSA Scholar Award from Yale Center for Clinical Investigation (all to Q.Y.), and NIH grant CA16359 (to the Yale Comprehensive Cancer Center). This publication was made possible by the CTSA Grant Number UL1 RR024139 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), and NIH roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCATS or NIH. The authors apologize to their colleagues whose work could not be cited due to space limitation.
Disclosure Statement
No competing financial interests exist.