
Abstract

The Reoviridae are nonenveloped, segmented, double-stranded RNA (dsRNA) viruses that include rotavirus, the most common cause of gastroenteritis in children (Parashar et al., 1998), bluetongue virus, an economically important pathogen of cattle and sheep, and mammalian orthoreovirus (reovirus), which infects humans, although disease is restricted to the very young (Dermody et al., In Press; Ouattara et al., 2011). Reovirus contains ten dsRNA segments packaged in two concentric protein shells, an outer capsid and core. During reovirus cell entry (Fig. 1), the outer capsid undergoes acid-dependent proteolysis catalyzed by cathepsin proteases, resulting in formation of infectious subvirion particles (ISVPs). ISVPs are characterized by the loss of viral capsid protein σ3 and cleavage of capsid protein μl into particle-associated fragments φ and δ. Further conformational rearrangements of the viral particle result in loss of the σ1 attachment protein and release of the transcriptionally active viral core into the cytoplasm via a penetration mechanism governed by the μl cleavage products (Chandran et al., 2002; Chandran et al., 2003; Odegard et al., 2004; Nibert et al., 2005; Danthi et al., 2008).
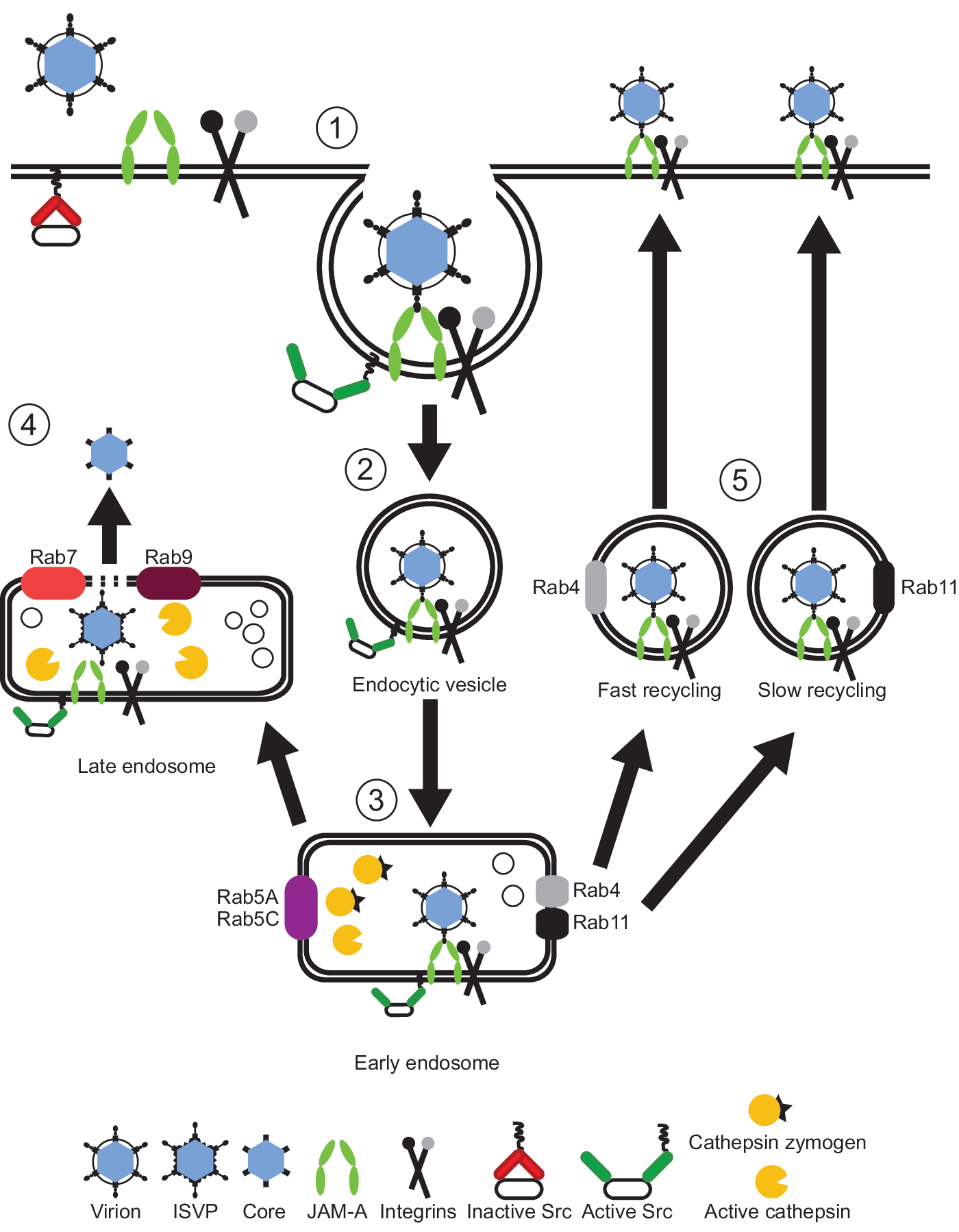
Model of reovirus cell entry. After attachment to cell-surface sialic acid and junctional adhesion molecule A (JAM-A), reovirus enters cells by receptor-mediated endocytosis (1) in a β1 integrin-dependent manner. Activation of Src kinase (2) at the site of entry targets reovirus to early endosomes (3), inhabited by inactive cathepsin proteases (cathepsin zymogens). In a productive infection, viral particles remain associated with JAM-A, and perhaps β1 integrin, and traverse to late endosomes where active cathepsin proteases mediate disassembly of viral particles, resulting in the release of the transcriptionally active viral core into the cytoplasm (4). In a nonproductive infection (5), viral particles traffic to fast and slow recycling compartments. The fate of viral particles within recycling endosomes is not known, but it is possible that these particles are redirected to the cell surface where they can reinitiate the entry process or disengage from receptors.
Reovirus binding to host cells is mediated by interactions of σ1 protein with cell-surface sialic acid and junctional adhesion molecule-A (JAM-A) (Barton et al., 2001a, 2001b; Forrest et al., 2003; Campbell et al., 2005). These receptors mediate reovirus attachment via an adhesion-strengthening mechanism in which low-affinity interactions with sialic acid tether virions to the cell surface before high-affinity binding to JAM-A (Barton et al., 2001a). However, JAM-A does not appear to participate in internalization of viral particles. The cytoplasmic tail of JAM-A, including a PDZ-binding domain that promotes the binding of JAM-A to scaffolding proteins in tight junctions (Severson and Parkos, 2009), is dispensable for reovirus infection (Danthi et al., 2006; Maginnis et al., 2006).
After receptor engagement, virions are internalized by receptor-mediated endocytosis in a manner dependent on β1 integrin (Maginnis et al., 2006; Maginnis et al., 2008). Uptake of virions is dependent on clathrin under certain conditions (Ehrlich et al., 2004; Maginnis et al., 2008), but there is mounting evidence that other mechanisms are involved in virion internalization (Maginnis et al., 2008; Schulz et al., 2012). It is not known whether β1 integrin mediates internalization of viral particles through a direct or indirect mechanism. However, viral core protein λ2 has solvent-exposed RGD and KGE integrin-binding motifs that could serve as β1 integrin contact residues. NPXY motifs in the cytoplasmic tail of β1 integrin are required for efficient internalization and infection by reovirus (Maginnis et al., 2008). Substitution of tyrosine residues with phenylalanines in the NPXY motifs leads to distribution of viral particles to degradative lysosomes (Maginnis et al., 2008). NPXY motifs are required for the association of β1 integrin with the actin cytoskeleton and also regulate integrin binding and signaling (Moser et al., 2009). Thus, it is possible that reovirus engagement of β1 integrin elicits recruitment of cellular molecules that promote internalization and proper endocytic sorting of reovirus particles.
After internalization, reovirus is transported to endosomal organelles where cathepsin proteases mediate stepwise proteolytic disassembly of virions (Ebert et al., 2002; Johnson et al., 2009; Doyle et al., 2012). Reovirus virions distribute to early, late, and recycling endosomal compartments during cell entry, but only those that traverse early and late endosomal compartments can establish infection (Mainou and Dermody, 2012). Particles that fail to induce proper signaling upon JAM-A and β1 integrin engagement may mis-sort into recycling endosomes and return to the cell surface, where they could reinternalize or float free. Within endosomes, JAM-A codistributes with reovirus virions during entry (Mainou and Dermody, 2012), suggesting that virus and receptor remain engaged after adsorption and are transported together to the site of viral disassembly. The fate of β1 integrin during reovirus endocytic transport is not clear.
Beyond the utilization of β1 integrin in reovirus internalization, the cellular molecules that promote uptake of viral particles and sorting within the endocytic pathway are beginning to be defined. Src kinase, the prototype member of the Src family of tyrosine kinases, colocalizes with reovirus particles during cell entry (Mainou and Dermody, 2011). Reovirus infection results in increased levels of Src kinase phosphorylation at tyrosine 416 (Mainou and Dermody, 2011), which is associated with increased kinase activity (Cooper et al., 1986; Parsons and Weber, 1989; Brown and Cooper, 1996). Furthermore, impairment of Src kinase activity with small-molecule inhibitors or decreased Src kinase expression using RNA interference leads to aberrant targeting of viral particles to lysosomes with a concomitant reduction in the efficiency of infection (Mainou and Dermody, 2011). It is possible that Src kinase mediates endosomal sorting of reovirus particles by recruiting and modifying molecules necessary for endocytic transport to a site of viral disassembly. Interestingly, other viruses use Src kinases for efficient cell entry.
Coxsackievirus, a member of the Picronaviridae, also utilizes Src family kinases during cell entry (Coyne and Bergelson, 2006). Coxsackievirus activates Abl and Fyn kinases after cell-surface engagement of decay accelerating factor (DAF). Abl kinase activation triggers Rac-dependent actin rearrangements that promote transport of viral particles and DAF to tight junctions. Fyn activation promotes caveolin phosphorylation and viral particle internalization using a caveolin-dependent pathway (Coyne and Bergelson, 2006). While reovirus likely accesses tight junctions to engage JAM-A, reovirus does not appear to induce actin rearrangement or use caveolin-dependent endocytic pathways during cell entry. As such, coxsackievirus and reovirus appear to use Src family kinases in distinct mechanisms to internalize into host cells.
The goal of reovirus endocytic transport is to gain access to an endocytic organelle replete with the enzymes capable of catalyzing virion disassembly. Cathepsins B, L, and S can mediate virion-to-ISVP disassembly in vitro and in cells (Ebert et al., 2002; Golden et al., 2004; Johnson et al., 2009; Doyle et al., 2012). Some cathepsin proteases (e.g., cathepsins B and L) are loaded into early endosomes in an inactive form, and after processing and acidification within endosomal organelles become active enzymes (Mohamed and Sloane, 2006; Guha and Padh, 2008). It is possible that multiple cathepsins catalyze disassembly during a single entry event. Cathepsins are active in a pH range of 4.5–8.0, depending on the specific cathepsin (e.g., the pH optimum is 5.0–6.0 for cathepsin B and 5.0–8.0 for cathepsin S) (Mohamed and Sloane, 2006; Guha and Padh, 2008). A gradient of cathepsin activity could provide a means for reovirus to regulate cathepsin-mediated disassembly by allowing specific cathepsins to act on the viral particle at distinct pHs as the virus traverses the endocytic pathway. Employment of multiple cathepsins for uncoating also may contribute to the broad tissue tropism and wide host range displayed by reovirus.
Several viruses, in addition to reovirus, exploit cathepsin proteases to achieve productive cell entry. After attachment and entry (Nanbo et al., 2010), Ebola virus is transported to late endosomes where cathepsins B and L cleave and activate the viral glycoprotein (Chandran et al., 2005; Misasi et al., 2012). The cleaved glycoprotein binds to Niemann-Pick C1, promoting fusion and entry into the cytoplasm (Carette et al., 2011; Cote et al., 2011). Henipaviruses also utilize cathepsins during cell entry. Hendra virus uses cathepsin L (Pager and Dutch, 2005), while Nipah virus uses cathepsins B and L (Pager et al., 2006; Diederich et al., 2012) to proteolytically process the F protein, a step required for fusion with target cells. However, in the case of these viruses, cathepsin-mediated cleavage occurs prior to viral release. Severe acute respiratory syndrome coronavirus (SARS-CoV) uses cathepsins to proteolytically process the viral spike glycoprotein before fusion with the cell membrane during entry (Simmons et al., 2005; Huang et al., 2006). Since cathepsins are required by several viruses for cell entry, it is possible that cathepsin inhibitors might serve as broadly active antiviral agents. Indeed, treatment of mice with a cathepsin L-specific small-molecule inhibitor decreases reovirus disease severity and mortality rate (Johnson et al., 2009).
Viruses use a variety of receptors and host molecules to target cells for replication and dissemination to new hosts. Viruses also exploit conserved cellular mechanisms, including kinase recruitment and activation, diminished pH, and cathepsin proteases to define specific locales within the cell to initiate endosomal egress. Further studies of pathogen uptake mechanisms will enhance an understanding of microbial pathogenesis and potentially illuminate new targets for anti-infective drug development.
Footnotes
Acknowledgments
We thank Caroline Lai, Jennifer Konopka, and Laura Ooms for critical review of the manuscript.
Disclosure Statement
No competing financial interests exist.