
Abstract
Recent studies are delineating a detailed picture of the architecture and function of the endoplasmic reticulum (ER) and the early secretory pathway, showing the existence of dynamic compartmentalization of ER quality control and ER-associated degradation (ERAD) factors. The compartmentalization is regulated by ER protein load and in turn regulates protein processing and cell fate. This compartmentalization is intimately linked to the protein quality control processes, protein disposal through ERAD, the unfolded protein response, and the initiation of apoptosis. It includes novel compartments, the ER-derived quality control compartment (ERQC), vesicles implicated in “ERAD-tuning,” and the mitochondria-associated membranes (MAMs).
Introduction
The ER-Derived Quality Control Compartment, the Calnexin Cycle, and the Mannose-Trimming Timer
In mammals, secretory proteins with slow folding or those that are terminally misfolded are segregated into a domain referred to as the ER-derived quality control compartment (ERQC) (Kamhi-Nesher et al., 2001). The relocation of proteins to this membrane-enclosed compartment involves microtubule-dependent trafficking (Kamhi-Nesher et al., 2001; Spiliotis et al., 2002; Wakana et al.; 2008) but is mostly unaffected by the drug brefeldin A, which causes ER–Golgi fusion (Kamhi-Nesher et al., 2001). The ERQC surrounds the centrosomes but is distinct from the Golgi, ERGIC, endosomes, and lysosomes also present in this region (Kamhi-Nesher et al., 2001, Spiliotis et al., 2002; Kondratyev et al., 2007).
Accumulation of unfolded or misfolded proteins is the trigger of ER stress, which activates the unfolded protein response (UPR) to restore the lost balance (Walter and Ron, 2011). Upon inhibition of their proteasomal degradation, or as a consequence of UPR induction, misfolded proteins accumulate in the ERQC together with the chaperone calnexin (CNX), and its soluble homolog, calreticulin (CRT), which are central components of the ER-folding and quality control machinery (Kamhi-Nesher et al., 2001; Spiliotis et al., 2002; Frenkel et al., 2004; Kondratyev et al., 2007; Tamura et al., 2010). UDP-glucose:glycoprotein glucosyltransferase (GT), which operates as the folding sensor in the CNX/CRT cycle (Benyair et al., 2011) reglucosylating N-glycans on incompletely folded glycoproteins for their binding to CNX/CRT (D'Alessio et al., 2010), was found in the ER exit sites (ERES) and peripheral ER (Cannon and Helenius, 1999) and does not accumulate in the ERQC (Kamhi-Nesher et al., 2001). CNX, in contrast, is absolutely excluded from the ERES (Cannon and Helenius, 1999). Like GT, glucosidase II—which removes terminal glucose residues from N-glycans, preventing reassociation of glycoproteins to CNX (D'Alessio et al., 2010)—appears to be in the peripheral ER (Lucocq et al., 1986). Normally, COPII coat proteins localize to the ERES (Budnik and Stephens, 2009), from which COPII-mediated vesicular transport of properly folded proteins initiates (Kuge et al., 1994; Pagano et al., 1999), reviewed in Benyair et al. (2011). However, a COPII component, p137, was found to relocate to the ERQC and colocalize with CNX upon adenosine triphosphate (ATP) depletion (Spiliotis et al., 2002).
Like GT, other ER-resident chaperones—the ER HSP70 BiP and the oxidoreductases protein disulfide isomerase (PDI) and ERp57 (Coe and Michalak, 2010)—are excluded from the ERQC (Kamhi-Nesher et al., 2001; Frenkel et al., 2004). CNX and CRT might function in the shuttling of misfolded proteins from the peripheral ER to the ERQC. The implication is that through the CNX/CRT cycle, slow-folding or misfolded glycoproteins would be segregated from the peripheral ER to the ERQC and then cycle back for deglucosylation, reglucosylation, and a new folding attempt (Fig. 1). During these cycles, the glycoproteins are exposed to the ERQC-localized ER mannosidase I (ERManI), which trims mannose residues on their N-glycans, tagging them for ER-associated degradation (ERAD) (Avezov et al., 2008; Hebert and Molinari, 2012). This slow trimming process is known as the mannose-trimming timer (Lederkremer and Glickman, 2005). High local concentration of ERManI in the ERQC could enable with time the production of extensively trimmed glycans (Hosokawa et al., 2003; Avezov et al., 2008) on terminally misfolded glycoproteins, which signals an end to their folding attempts. ERManI was also reported to appear at or near the Golgi (Pan et al., 2011), and a proteomics study located it mainly near COPI vesicles (Gilchrist et al., 2006). Our recent unpublished results indicate that ERManI actively cycles between the ERQC (where it encounters its substrates) and other pre-Golgi compartments also in undisturbed conditions. N-glycan trimming by ERManI was shown to be an essential step required for glycoprotein accumulation and retention in the ERQC (Avezov et al., 2008; Groisman et al., 2011).
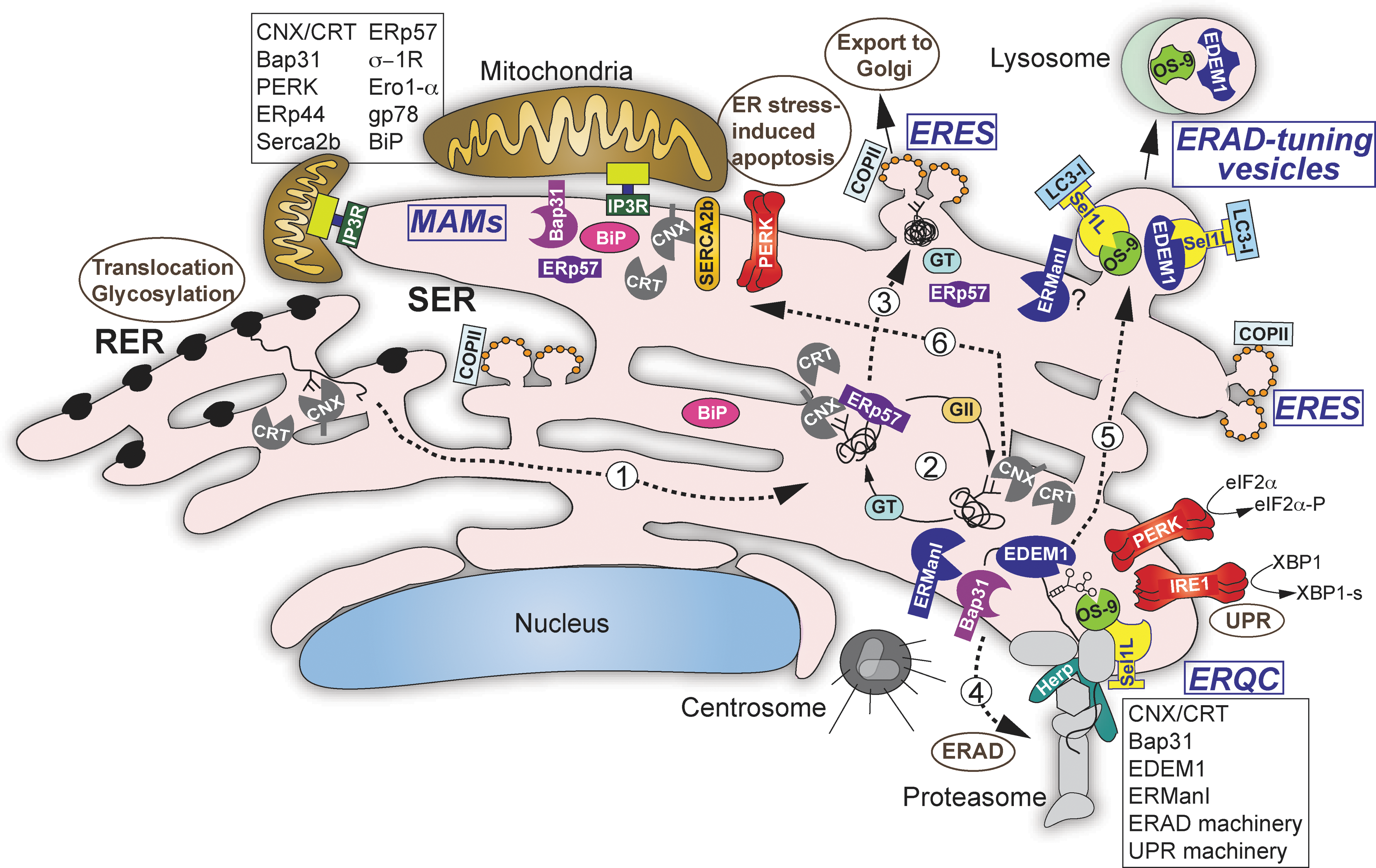
Model of compartmentalization of ER quality control and ERAD factors in mammalian cells. A protein is cotranslationally translocated into the lumen of the rough endoplasmic reticulum (ER), N-glycosylated, and the two terminal glucose residues are excised from the sugar chain precursor resulting in Glc1Man9GlcNAc2, which is recognized by calnexin (CNX) or calreticulin (CRT; step 1). This starts the CNX folding cycle (step 2), which involves the oxidoreductase ERp57 for disulfide bond formation, GII for removal of the glycoprotein from the folding attempts by trimming the final glucose residue from its sugar chain, and the folding sensor GT, which reglucosylates the glycoprotein returning it to the CNX cycle. The glycoprotein is shuttled between the peripheral smooth ER and the juxtanuclear pericentriolar ERQC, possibly with the aid of CNX/CRT or Bap31. Upon successful folding, the glycoprotein is released from the CNX cycle and is transported to ER exit sites (ERES) where it is packaged into COPII vesicles and transported to the Golgi (step 3). If after several cycles the glycoprotein cannot fold properly it is further trimmed (by ERManI and/or EDEMs 1-3), leading to its release from the CNX cycle because of the removal of the acceptor mannose for glucose transfer by glycoprotein glucosyltransferase (GT). After extensive trimming of its glycans, the glycoprotein is recognized by the OS-9 or XTP3B lectins and targeted to ERAD (step 4), where the glycoprotein is ubiquitinated by the HRD1-SEL1L complex (or other ubiquitin ligases) and sent to proteasomal degradation (step 4). Unglycosylated misfolded proteins are also targeted to the ERQC and ERAD by a less clear mechanism. Herp is a key factor in the dynamic organization of the ERAD complex at the ERQC. SEL1L is proposed as the receptor for segregation of short-lived components (e.g., EDEM1 and OS-9) into LC3-I–coated ERAD-tuning vesicles (step 5). These components are finally degraded in lysosomes. Upon ER stress, the UPR sensors PERK and IRE1 also accumulate at the ERQC, which enables their physical separation from BiP and UPR amplification. Prolonged ER stress causes rapid calcium transfer from the ER to mitochondria through IP3Rs channels at the mitochondria-associated membranes (MAMs), initiating an apoptotic cascade. A series of ER factors are recruited to the MAMs, inducing the apoptotic pathway, like PERK and Bap31; some affect IP3R activity (e.g., BiP and Ero1α), while others affect the SERCA2b calcium pump (CNX) (step 6). For simplicity, several accessory factors have not been included. ERQC, ER-derived quality control compartment; ERAD, ER-associated degradation; UPR, unfolded protein response; PKR, protein kinase RNA; PERK, PKR-like ER kinase; IRE1, inositol-requiring enzyme-1; EDEM1, ER degradation-enhancing α mannosidase-like protein 1; RER, rough ER.
Misfolded glycoproteins also associate with ER degradation-enhancing α mannosidase-like protein 1 (EDEM1), suggested to participate in the trimming of their sugar chains (Olivari et al., 2006; Hosokawa et al., 2010). EDEM1 is recruited to the ERQC under ER stress (Groisman et al., 2011; Walter and Ron, 2011), when its expression increases significantly (Yoshida et al., 2003). At these high UPR-induced levels, EDEM1 overrides the requirement for mannose trimming, accelerating ERAD of both glycosylated and nonglycosylated substrates (Ron et al., 2011; Marin et al., 2012). Thus, high local concentration of EDEM1 in the ERQC upon ER stress enables effective but less specific protein disposal, promptly clearing the secretory pathway.
Compartmentalization of ERAD Machinery
EDEM1 targets the misfolded glycoproteins to the receptor lectins OS-9 and XTP3-B (Hosokawa et al., 2009; Lederkremer, 2009; Groisman et al., 2011). Both OS-9 and XTP3-B deliver the misfolded glycoproteins to the HRD1-SEL1L membrane-anchored ubiquitin ligase complex for ERAD (Christianson et al., 2008; Hosokawa et al., 2008) at the ERQC (Kondratyev et al., 2007; Groisman et al., 2011). Interestingly, similar to ERManI, OS-9 was found to localize in the ERQC without requiring misfolded protein accumulation (Avezov et al., 2008; Ron et al., 2011).
Additional ERAD machinery components shown to be recruited to the ERQC upon ER stress include membrane-bound Derlin-1 and soluble E3 ligase SCFFbs2, AAA ATPase p97/VCP, proteasomes, and ubiquitinated proteins, which associate with the ERQC membranes on its cytosolic side (Kondratyev et al., 2007; Groisman et al., 2011) (Fig. 1). Derlin-1 is recruited to a large complex, together with HRD1-SEL1L and p97/VCP (Ye et al., 2003), which possibly functions in retrotranslocation (Wahlman et al., 2007). Sec61β (Schmitz et al., 2004) and Bap31 (Wang et al., 2008) are recruited to the ERQC as well (Kamhi-Nesher et al., 2001; Wakana et al., 2008) and were also suggested to be involved in the retrotranslocation process, although no consensus exists yet for their participation. In conclusion, some early quality control factors remain in the peripheral ER segregated from late quality control and ERAD components that, upon demand, when misfolded proteins accumulate, are recruited to the ERQC, enabling separation of the processes of folding and targeting to degradation in space and time (Table 1). Studies in S. cerevisiae suggest that a similar compartmentalization might also exist in yeast, where an ER-associated compartment (ERAC) was proposed as a quality control subcompartment for misfolded protein retention and targeting to ERAD (Huyer et al., 2004).
EDEM1, ER degradation-enhancing α mannosidase-like protein 1; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ERQC, ER-derived quality control compartment; CNX, calnexin; CRT, calreticulin; IRE1, inositol-requiring enzyme-1; PDI, protein disulfide isomerase; PERK, PKR-like ER kinase; PKR, protein kinase RNA; UPR, unfolded protein response.
Targeting of Short-Lived Machinery Components—ERAD Tuning
Unlike many of the long-lived ER chaperones, some quality control and ERAD components (e.g. EDEM1, ERManI, OS-9, XTP3B, Herp, HRD1, SEL1L) are short-lived. These factors participate in the targeting to ERAD, and in order to avoid interference with folding attempts of newly synthesized proteins (Avezov et al., 2008; Alcock and Swanton, 2009; Ron et al., 2011), they are kept at low levels. As we have mentioned for EDEM1, and we will see below for Herp, the levels of some of these proteins is highly regulated and increased upon demand of ER protein load. The proteasomes are in charge of the fast turnover of some of them (e.g., Herp, HRD1, SEL1L) (Hori et al., 2004; Iida et al., 2011), whereas the degradation of others (e.g., EDEM1, ERManI, OS-9) is also mediated by lysosomes. It was observed that in resting cells, at the steady state, EDEM1 exists in vesicles that are segregated from the ER (Zuber et al., 2007). EDEM1 and OS-9 were shown to be selectively removed from the ER by LC3-I–coated vesicles named EDEMosomes, distinguished from the LC3-II–covered autophagosomes and from COP-II–coated secretory vesicles (Cali et al., 2008; Reggiori et al., 2010). This process is called ERAD tuning (Bernasconi and Molinari, 2011). SEL1L was suggested to serve as a selective ERAD-tuning, regulating receptor for EDEM1 and OS-9 segregation, as it associates to these proteins and also to LC3-I (Bernasconi et al., 2012). This association is inhibited upon expression of misfolded polypeptides. ERAD machinery segregation in the ERQC and ERAD tuning are both mechanisms that protect proteins that are still incompletely folded from unwanted recognition by the ERAD machinery.
Compartmentalization of the UPR
As mentioned above, the abundant ER chaperone BiP, implicated in protein folding and activation of the UPR (Walter and Ron, 2011), was shown to be excluded from the ERQC even under stress conditions (Kamhi-Nesher et al., 2001; Kondratyev et al., 2007). Consistently, a large complex of chaperones bound to BiP did not contain the ERQC-targeted CNX or CRT (Meunier et al., 2002).
Although BiP remains distributed through the ER, the UPR sensors—protein kinase RNA (PKR)-like ER kinase (PERK) and inositol-requiring enzyme-1 (IRE1)—are recruited to the ERQC upon accumulation of misfolded proteins (Kondratyev et al., 2007). This physical separation of BiP from PERK and IRE1 would contribute to the activation of the UPR. Paradoxically, PERK-mediated phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) is both necessary and sufficient for ERAD machinery and substrate accumulation at the ERQC (Kondratyev et al., 2007). Phosphorylated eIF2α itself is targeted to the cytosolic side of the ERQC and, in a positive feedback loop, causes protein concentration at the compartment, which in turn would amplify the UPR (Kondratyev et al., 2007). A membrane-bound ERAD component, homocysteine-induced endoplasmic reticulum protein (Herp) (Schulze et al., 2005; Okuda-Shimizu and Hendershot, 2007), which is highly UPR-induced downstream of eIF2α phosphorylation (van Laar et al., 2000; Kokame et al., 2000; Ma and Hendershot, 2004), seems to be responsible for the recruitment of ERAD machinery to the ERQC (our unpublished results). Furthermore, Herp itself was found to redistribute to the ERQC following ER stress. Thus, being regulated by the UPR and recruiting ERAD components, Herp is proposed to coordinate the processes of UPR and ERAD by organizing a functional ERAD complex at the ERQC.
Similar to the concentration of UPR sensors in the ERQC, it was shown that upon UPR induction in yeast, the IRE1 protein and HAC1 mRNA (the yeast homolog of XBP1), were found in discreet clusters, which was vital for UPR signaling (Aragon et al., 2009).
Prolonged ER stress causes a rapid transfer of calcium from the ER to the mitochondria, eliciting the induction of the mitochondrial apoptotic pathway (Boehning et al., 2003; Simmen et al., 2005). Exchange of calcium (and also of lipids) between the two organelles (Raturi and Simmen, 2012) takes place at distinct domains termed mitochondria-associated membranes (MAMs) (Vance, 1990). The Ca2+ exchange is mainly through the inositol 1,4,5-trisphosphate receptors (IP3Rs), which are ligand-gated calcium channels that accumulate at the MAMs (especially type 3 IP3Rs) (Mendes et al., 2005). There are several IP3R interactors and regulators, for example, the molecular chaperone σ-1 receptor, BiP, and the oxidoreductases Ero1α and ERp44 (Hayashi et al., 2009; Li et al., 2009; Higo et al., 2010). The induced or impaired association of IP3Rs with these interactors at the MAMs following ER stress has been proposed to regulate the onset of the apoptotic pathway (Li et al., 2009; Higo et al., 2010). Several anti- and pro-apoptotic BCL-2 family members also regulate IP3R function (Rodriguez et al., 2011). Other components of ER quality control and ERAD machineries that are recruited to the MAMs have also been suggested to play a role in the regulation of Ca2+ release—among them CNX, CRT, ERp57, and gp78 (Li et al., 2006; Hayashi et al., 2009). CNX undergoes palmitoylation, which was proposed to regulate its traffic to the MAMs (Lynes et al., 2012). At the MAMs, CNX associates with the sarcoendoplasmic reticulum calcium ATPase (SERCA) 2b regulating its Ca2+ transport (Roderick et al., 2000; Myhill et al., 2008).
Additional pathways have been proposed for induction of apoptosis at the MAMs. For instance, PERK was recently reported to accumulate at the MAMs where it is required to induce apoptosis following reactive oxygen species (ROS)-based ER stress (Verfaillie et al., 2012). Another pathway implicates interaction at the MAMs of mitochondrial fission protein (Fis1) with Bap31, which facilitates the cleavage into the pro-apoptotic p20Bap31 and the subsequent recruitment of Procaspase-8 (Iwasawa et al., 2011). Since both Bap31 and CNX were found to cycle between ER subdomains, including the rough ER, the ERQC, and the MAMs (Myhill et al., 2008; Wakana et al., 2008; Iwasawa et al., 2011), they might play a role as mediators of signaling between the ER subcompartments.
In summary, the compartmentalization of ER quality control and ERAD factors is a dynamic process regulated by secretory protein load and which, in turn, regulates protein processing and cell fate. As ER protein load increases, many factors that are normally distributed throughout the ER or segregated into vesicles congregate at the ERQC, organizing the quality control and ERAD machineries. High levels of protein load and ER stress divert some of these factors to the MAMs, initiating apoptosis.
Footnotes
Acknowledgments
We apologize to authors whose work we were unable to cite owing to space limitations. Related work was supported by a grant from the Israel Science Foundation (1070/10).
Disclosure Statement
No competing financial interests exist.