
Abstract
p62 is a multidomain protein that contains different kinds of protein–protein interaction domains, including an N-terminal PB1 domain, a ZZ-type zinc finger domain, a nuclear localization signal (NLS), an export motif (NES), the LC3-interacting region (LIR), the KEAP1-interacting region (KIR), and a C-terminal Ub-associated domain (UBA). p62 is involved in the degradation of protein aggregates and cytoplasmic bodies via selective autophagy through its PB1, LIR, and UBA domains to maintain homeostasis in the cell. Moreover, NES, NLS, KIR, and ZZ domains have been found to be linked to ubiquitinated protein degradation by autophagy. Therefore, understanding the functional domains of p62 is important. In this review, we attempt to expound the mechanism of connection between p62 and selective autophagy to illustrate how the domains of p62 regulate selective autophagy, and to provide a new direction and perspective on selective autophagy research.
Introduction
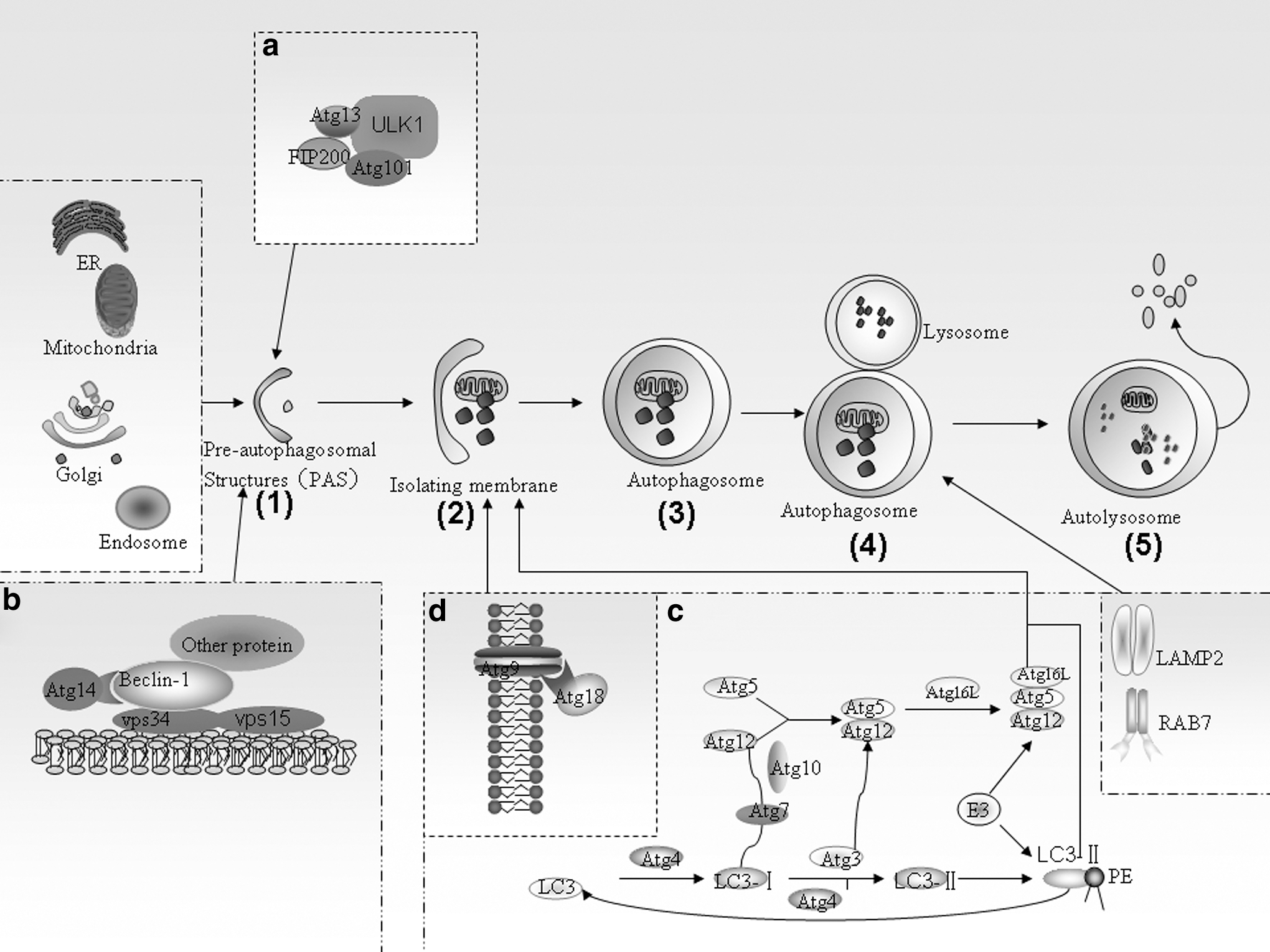
The process of autophagy. Autophagosome formation is initiated by
Autophagy was originally considered a nonselective bulk degradation process, However, the ability of autophagosomes to degrade substrates in a selective manner has now been clearly established (Johansen and Lamark, 2011; Shaid et al., 2013). Mechanistic insights into selective autophagy have been provided by studies on p62, an autophagy-specific substrate that functions as an autophagic adapter in mammals. It can sequester polyubiquitinated proteins and polymerize them into a cytoplasmic storage before their degradation. Recent studies have revealed that p62 directs autophagic uptake of proteins via its LIR motif and subsequently forms protein bodies that contain LC3 and are degraded by autophagy (Bjorkoy et al., 2005; Pankiv et al., 2007). In this manner, p62 protects the cytosol from the toxic effects of misfolded or mutated proteins (Bjorkoy et al., 2005; Gao and Chen, 2011). In addition to ubiquitinated protein aggregates, peroxisomes labeled with ubiquitinated proteins are also degraded by autophagy in a p62-dependent manner (Kim et al., 2008). Furthermore, p62 has been found to interact with atypical PKCs (PKCζ and PKCι/λ) in several signaling pathways and to participate in receptor-mediated signal transduction (Moscat and Diaz-Meco, 2000; Jiang et al., 2009).
In addition to the LIR motif, p62 has several other interesting protein–protein interaction motifs: PB1 in its N-terminal region that binds atypical PKCs; UBA in its C-terminal region that interacts with ubiquitinated proteins marked for proteasomal degradation; ZZ finger, a binding site for the RING finger protein tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6); and NLS, NES, and two PEST sequences for rapid protein degradation (Geetha and Wooten, 2002; Bjorkoy et al., 2006) (Fig. 2). The presence of these different functional domains suggests that p62 plays pleiotropic roles in the regulation of cellular signaling and homeostasis of multiple proteins. Some new studies have reported that p62 is required for shuttling the microtubule-binding protein Tau for autophagic degradation via its UBA domain, and disruption of the p62 gene in mice results in an Alzheimer's disease (AD) phenotype (Ramesh et al., 2008; King et al., 2009). Moreover, targeted deletion of mice p62 have been found to lead to insulin resistance, type 2 diabetes, and leptin resistance (Rodriguez et al., 2006; Moscat and Diaz-Meco, 2011). However, the identities of the specific p62 domains involved in these diseases are unknown. Further understanding the relationship between p62 and autophagy and the functional domains involved in p62-regulated autophagy has significant implications for tackling these diseases. This review aims to provide a detailed account of the functions of various p62 domains known to date in selective autophagy.
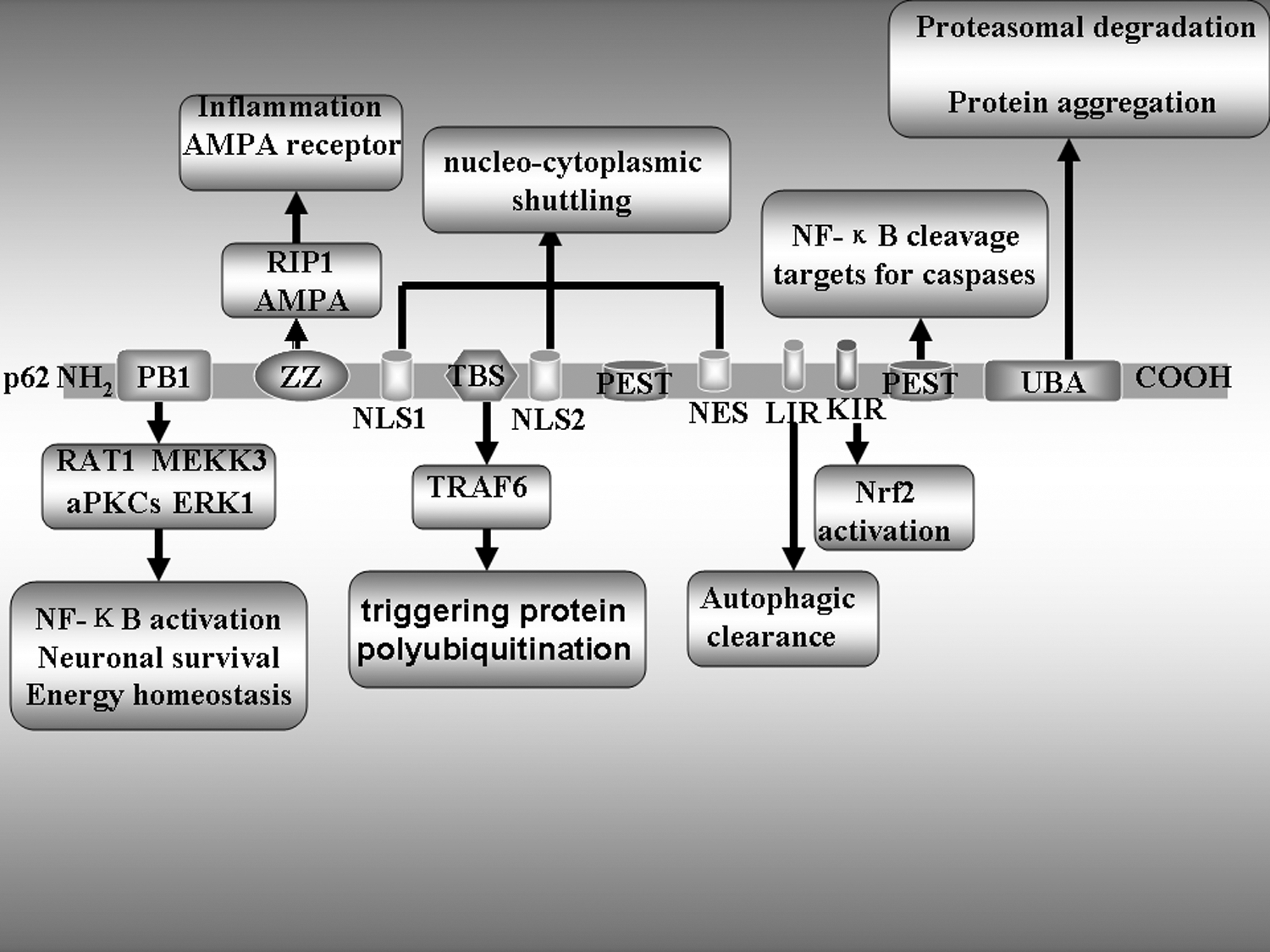
The functional domains of the p62 protein. The figure depicts different protein–protein interaction motifs and their location in the p62 protein. The most important functional interactions of these domains have been identified. Domain organization and interacting partners of p62. PB1 and Phox/Bem domain 1 interact with ERK1 to control adipogenesis. Together with aPKCs, they control NF-κB. The interaction with NBR1 also occurs through PB1, but its role needs to be clarified. ZZ, atypical zinc finger, governs the interaction with RIP and may be relevant for tumor necrosis factor-activated NF-B. TB, TRAF6 binding, accounts for p62's role in IL-1, NGF, and RANK toward NF-B. LIR, LC3-interacting region, locates p62 in the autophagosomes. KIR, Keap-interacting region, regulates NRF2 activation. UBA, ubiquitin-associated, mediates the interaction with polyubiquitinated proteins, including caspase-8, and modulates TRAF6 interaction and activity. PEST, is rich in proline(P), glutamic acid(E), serine(S), and threonine(T), modulates NF-κB cleavage and targets for caspases.
LIR-ATG8 (LC3/GABARAP) Interaction: The Underlying Connection Between p62 and Selective Autophagy
The LC3-interacting region (LIR) of p62 motif that consists of 11 amino acids that mediate the interaction of p62 with the LC3/GABARAP family of proteins for autophagic degradation (Ichimura et al., 2008b). Pankiv et al. (2007) first mapped the LIR of p62 to the region encompassing amino acids 321 to 342 of human p62, which contains an evolutionarily conserved motif of three acidic residues. Mutation of these three aspartic acid residues to alanine or a single mutation of W to A strongly affects binding to LC3 (Noda et al., 2008). Soon after, Ichimura et al. (2008b) identified a specific region of p62 consisting of 11 conserved acidic and hydrophobic residues (Ser334 to Ser344) as the LC3 recognition sequence (LRS). This opened the door for determining the crystal structure of the LC3-LRS complex. This amino acid sequence is almost identical to a previously reported LIR of human p62. Komatsu and Inagaki's group subsequently determined the structures of the LC3–p62 peptide via X-ray and nuclear magnetic resonance crystallography (Ichimura et al., 2008b). The LIR of p62 adopts an extended β-conformation and forms an intermolecular parallel β-sheet with β2 of LC3 that relies on the core sequence DDDWTHL of p62 LIR. LIR is located in the interface between the N-terminal arm and the C-terminal of the Ub-like domain of the Atg8 protein. The side chains of the W and L sites are blind to two hydrophobic pockets in the Ub-like domain of LC3 (Pankiv et al., 2007; Noda et al., 2008, 2010).
Aside from two hydrophobic interactions, the p62-LC3 interaction also requires three consecutive aspartate residues (DDD) of the LIR motif and the N-terminal arm and the Ub-like domain of LC3. Moreover, functional requirement of both the WXXL core motif and the acidic residues of the p62 LIR motif have been verified by substitution of these residues with alanine (Johansen and Lamark, 2011; Ichimura et al., 2008a). The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 for autophagic degradation, as demonstrated by the Shvets's group (Shvets et al., 2008).
The LIR domain has been identified in other ATG8 interactors, including Atg19, NBR1, Nix, calreticulin, and clathrin (Mohrluder et al., 2007; Kirkin et al., 2009; Novak et al., 2010). Recently, Pankiv et al. (2010) reported that a novel protein of FYCO1 (FYVE and coiled-coil domain-containing protein 1) binds to LC3, Rab7, and PtdIns-3-phosphate on autophagosomes, endosomes, and lysosomes. FYCO1–LC3 interacts via the LIR motif adjacent to the FYVE domain of FYCO1. The FYCO1–LC3 complex on the autophagosomes promotes kinesin-mediated microtubule plus end-directed transport of autolysosomes. In addition, FYCO1 could be recruited to the outer membrane of autophagosomes, it is not degraded by autophagy (Pankiv et al., 2010; Pankiv and Johansen, 2010). A study found that disheveled (Dvl), an important adaptor protein in the Wnt-signaling pathway, interacts with LC3 via an LIR motif and is degraded by autophagy (Gao and Chen, 2011).
The p62 LIR motif has an important function in selective autophagy, it influences cargo transport in the autophagy process. However, merely relying on the LIR motif is not sufficient to degrade a protein by selective autophagy. Analogous to the oligomerization and aggregation of preApe1 via the Cvt pathway in yeast. Recently, Eisuke and Noboru reported that the PB1 domain can mediate oligomerization of p62 in the ER, but the LIR motif is not involved in the process (Itakura and Mizushima, 2011). In addition, they proposed that oligomerization of p62 is required for the formation of autophagosomes (Itakura and Mizushima, 2011). Thus, p62 is required in forming p62 bodies, which are subsequently degraded by autophagy, the LIR motif and PB1 domain also require the UBA domain that binds to Ub-labeled proteins (Bjorkoy et al., 2005; Tung et al., 2010)
UBA Domains and p62-Mediated Aggresome Formation
The C-terminal UBA domain of p62 is composed of a three-helix bundle of 50 amino acids. Since it contains Lys63-linked Ub chains that have a more open and extended conformation than Lys48-linked Ub chains, the UBA domain preferentially binds to Lys63-linked polyubiquitin chains (Long et al., 2008). p62, ubiquilin-1, and NBR1 have UBA-type binding motifs (Li et al., 2007; Waters et al., 2009). The most important UBA domain of p62 can bind to polyubiquitinated proteins and shuttle them for proteasomal degradation. However, the specificity of this p62-UBA domain-mediated transport requires further clarification. For instance, tau proteins in neurons are transferred to proteasomes by p62 (Ramesh et al., 2008). Hocking et al. (2002) and Layfield and Hocking (2004) demonstrated that mutations in the UBA domain are linked to Paget's bone disease. This could be attributed to p62 mutations that increase osteoclastogenesis by potentiating receptor activator of nuclear factor-kappaB ligand (RANKL)-induced activation of nuclear factor-kappaB (NF-κB). TRAF6 and/or the atypical protein kinase Cζ (PKCζ) may be involved in this process and selective autophagy may regulate RANKL signaling (Layfield et al., 2004; Yip et al., 2006; Ang et al., 2009). Mutations in the UBA domain of p62 can thus predispose one to the development of Paget's bone disease by disrupting proteasomal targeting of proteins, aggregate formation, and autophagic clearance, but do not cause the disease (Fig. 2).
Several studies have demonstrated that p62 has a crucial role in the formation of ubiquitinated protein aggregates within cells (Bjorkoy et al., 2005, 2006; Kraft et al., 2010). Although many studies have reported that overexpression of p62 enhances the formation of polyubiquitinated aggregates, the molecular mechanism of aggregate formation is still unknown. Paine et al. (2005) demonstrated that overexpression of p62 can lead to their self-aggregation. The UBA domain mediates the accumulation of p62 proteins into aggresomes or aggresome-like inclusions with ubiquitinated misfolded proteins, also called p62 bodies. Bjorkoy et al. (2006) demonstrated that protein aggregates containing the p62 protein can be cleared out via autophagic uptake. In addition to p62, Nezis et al. (2008) reported that refractory to Sigma P [Ref(2)P], the melanogaster p62 homologue, which induced protein aggregates in the adult brain by a reduced autophagic or proteasomal activity or melanogaster models of human neurodegenerative diseases through its UBA and PB1 domain.
PB1 Domain: The Foundation of p62 as a Signaling Scaffold Molecule
The PB1 (Phox and Bem1p) domain in the N-terminus of p62 is a crucial evolutionarily conserved dimerization/oligomerization domain that organizes homodimers and heterodimers (Lamark et al., 2003; Noda et al., 2003; Wilson et al., 2003). p62 is involved in different pathophysiological processes, such as osteoclastogenesis, angiogenesis, and early cardiovascular development or cell polarity, via its PB1 domain (Hirano and Inagaki, 2006). Some studies have found that the PB1 domain can interact with PKCζ, PKCι/λ, MEKK3, MEK5, and ERK1, which contain the corresponding PB1 motifs (Fig. 2) and regulate different signaling pathways (Wilson et al., 2003; Nakamura et al., 2010). Moreover, p62 stimulates NF-κB signaling by forming complexes with atypical PKCζ. This signaling activation is needed upstream of the kinase RIP. The efficiency of NF-κB signaling induced by IL-1 and TNF-α via the p62-PKCζ complex can be enhanced by the binding of Ajuba to this complex (Feng and Longmore, 2005).
The p62-MEKK3 complex binds TRAF6, which regulates the ubiquitination of the IKK complex and the activation of NF-κB. Furthermore, the p62/PKCζ complex can bind the ß-subunit of the delayed rectifier K+ channel (Kvβ) and regulate the function of Kv channels that induce neuronal hyperpolarization. Some studies have demonstrated that norepinephrine and a1-adrenoceptor agonist can increase the expression of the p62 protein and induce hyperpolarization in PC12 cells (Kim et al., 2005). In addition, the self-interaction of p62 is important because it gives rise to p62 bodies/ALIS and is required for the efficient degradation of p62 by selective autophagy (Paine et al., 2005). The PB1 domain of p62 interacts with the proteasomal Rpt-1 protein and lipid droplets and triggers the degradation of ubiquitinated p62 cargo proteins and other substances via the 26S proteasome complex (Seibenhener et al., 2004). For instance, the ubiquitinated tau protein can be shuttled to proteasomal degradation via the p62 carrier (Babu et al., 2005). These data indicate that p62 is a multifunctional scaffold protein that serves a large variety of cellular functions via its PB1 domain.
ZZ Domain: A Mysterious Domain of p62
Some studies have found that the ZZ-type zinc finger domain of p62 binds to the RIP1 protein and links PKCζ to form a signaling complex (Festjens et al., 2007). The death domain of RIP1 kinase interacts with the TNF receptor, and this complex can activate the NF-κB and p38MAPK signaling pathways (Yu et al., 2009). Yu et al. (2009) reported that the IDR-1 peptide can bind to the ZZ domain of p62 and clearly enhance the formation of the RIP1 complex, but not those of p62 and PKCζ in TNF-α-treated cells. IDR-1 strongly activates signaling via the p38-C/EBPb pathway and inhibits inflammation. The function of the ZZ domain of p62 in the regulation of cellular signaling is still unclear. Recently, Jiang et al. (2009) demonstrated that the ZZ domain of p62 could directly interact with AMPA receptor subunits and influence their trafficking and phosphorylation. They also observed that the cell surface level of GluR1 and GluR1 phosphorylation status are clearly reduced in mice that lack p62, although no difference was found in the total expression of GluR1 and NMDA receptors compared with the control mice. The deficiency in AMPA receptor trafficking to synaptic membranes most likely contributes to the decline in LTP observed in p62 knockout mice (Jiang et al., 2009). Thus, p62 can regulate synaptic plasticity by enhancing AMPA receptor translocation (Fig. 2).
TBS and KIR Domains
In addition to the PB1 and ZZ domains, a TRAF6-binding domain (TBS) mediates the binding of p62 to TRAF6, an E3 ubiquitin (Ub) ligase that triggers protein polyubiquitination (Jadhav et al., 2008, 2011). Several studies have demonstrated that this may also occur via Kelch-like ECH-associated protein 1 (Keap1), which is a component of the cullin-3-based E3 ubiquitin ligase and is responsible for the recognition of ubiquitination substrates, such as NRF2 (Fan et al., 2010; Johansen and Lamark, 2011). Ub ligases are present in most protein inclusions, and the Ub chains present in mature p62 bodies or aggresomes may differ from the Ub chains initially used to recruit p62 or HDAC6 (Lee et al., 2010). Experimentally distinguishing the role of Ub in recognition versus its role in the construction of autophagy-competent structures is difficult (Lee et al., 2010). Some new studies have revealed that p62 interacts with Keap1 via a motif designated in the Keap1 interacting region (KIR) and this interaction blocks the binding between Keap1 and NRF2, dislodges NRF2 from interacting with Keap1, and stabilizes NRF2 (Nguyen et al., 2009; Jain et al., 2010; Komatsu et al., 2010). Thus, overproduction of p62 or deficiency in autophagy competes with the interaction between Nrf2 and Keap1, resulting in stabilization of Nrf2 and transcriptional activation of Nrf2 target genes (Fig. 2).
NLS and NES Domains
p62 has generally been considered a cytosolic protein, and little attention has been paid to its possible nuclear functions. Pankiv et al. (2010) found that p62 continuously shuttles between nuclear and cytosolic compartments using two nuclear localization signal domains (NLS1 and NLS2) and one nuclear export motif (NES) in the structure of p62. They also demonstrated that p62 can undergo rapid nucleo-cytoplasmic shuttling, which is regulated by the phosphorylation of p62 close to the NLS2 site (Pankiv et al., 2010), and inhibition of nuclear export leads to accumulation of ubiquitinated proteins in the nucleus and further accelerates trafficking of p62 into the nucleus (Pankiv et al., 2010). Interestingly, p62 and ubiquitinated proteins colocalize in promyelocytic leukemia, a nuclear proteasomal center (Wang et al., 2011). Furthermore, several studies have shown that the p62 protein can be localized in nuclear aggregates (Pikkarainen et al., 2008, 2011). Taken together, these studies indicate that p62 is also involved in the proteasomal degradation of ubiquitinated proteins in the nucleus via its NLS and NES domains.
PEST Sequences: The Basis for Targeting of Proteins for Degradation by p62
The PEST sequence is a peptide sequence rich in proline (P), glutamic acid (E), serine (S), and threonine (T) (Rechsteiner and Rogers, 1996). PEST sequences in proteins function as proteolytic signals for rapid degradation, leading to short intracellular half-lives. For instance, calpain recognizes the PEST and cleaves it (Pursiheimo et al., 2009). In addition, c-myc and Notch-1, two short-lived proteins, are are also known to be ubiquitinated by the E3 ligase and degraded by the proteasome pathway based on their PEST sequences. HS-1-associated protein X-1 (H) (Hax-1) is also a short-lived protein that is rapidly degraded by proteasomes based on its PEST sequence, and this capacity for quick degradation may underlie rapid cellular responses to different stimulations. Similarly, p62 is upregulated in cancerous cells and in response to various stimuli (Li et al., 2012). p62 could be rapidly degraded by the autophagic-lysosomal system especially in hypoxic conditions (Pursiheimo et al., 2009). However, the mechanisms by which PEST sequences contribute to the regulation of p62 function are not completely clear. In addition, PEST sequences are also targets of caspases. p62 can be cleaved by caspase-6 and caspase-8, resulting in inhibition of the autophagic clearance of ubiquitinated proteins (Belizario et al., 2008).
Autophosphorylation of p62: A New Function That Regulates Selective Autophagy
p62 is an adaptor or scaffold protein that participates in the activation of the transcription factor NF-κB by binding with aPKC, RIP1 kinase, or TRAF6 during tumorigenesis and osteoclastogenesis. p62 is also a key molecule that manages autophagic clearance of polyubiquitinated proteins. As p62 itself is degraded during selective autophagy, p62 proteins that mediate signal transduction should be distinct from those that manages selective autophagy. However, how p62 controls autophagic degradation of polyubiquitinated proteins remains unclear.
Matsumoto et al. (2011) showed that, under normal conditions, p62 phosphorylation is regulated by CK. Once the proteasome pathway is inhibited or overloaded, polyubiquitinated proteins (polyUb proteins) are accumulated, and specific phosphorylation of p62 at serine 403 (S403) [S403-phos-p62] occurs, which binds to the accumulated Ub proteins. The binding of S403-phos-p62 to Ub proteins is dephosphorylation resistant. Then, the S403-phosphorylation balance shifts to an increased S403-phos-p62 state and S403-phos-p62 forms sequestosomes with Ub proteins (Matsumoto et al., 2011). An isolation membrane is recruited to the sequestosome by the LC3–p62 interaction and the autophagosome grows on the surface of sequestosomes (Matsumoto et al., 2011). Eventually, autophagosomes holding sequestosomes fuse with lysosomes and undergo degradation.
Accumulation of ubiquitinated protein inclusions is a characteristic of several neurodegenerative diseases. Selective autophagy of the ubiquitinated protein is enhanced by S403 phosphorylation of p62, and this enhancement reduces the accumulation of protein aggregates. Thus, p62 phosphorylation at S403 is an appropriate therapeutic target for the induction of autophagic degradation of pathogenic ubiquitinated proteins in neurodegenerative diseases.
Concluding Remarks and Future Directions
p62 is a cellular “Swiss Army Knife,” with diverse functions in the cell arising from its unique functional motifs and protein–protein interaction properties (Nezis and Stenmark, 2012). p62 has a significant function in the regulation of oxidative stress signaling, neurodegenerative diseases, and tumorigenesis. Furthermore, p62 is a key regulator of nutrient sensing in the mTORC1 pathway and has important implications for the deregulation of the mTORC1 cascade in cancer (Nezis and Stenmark, 2012). It has been found that p62 senses nutrient signals and activates mTORC1, thus inhibiting autophagy and creating a loop that results in enhanced p62 levels (Moscat and Diaz-Meco, 2011). High-calorie diets promote lipogenesis and adiposity through mTORC1 activation, both of which are antagonized by p62. p62 itself is modulated by autophagy, which likely controls the anti-inflammatory actions of PKCξ. In recent years, several studies have highlighted p62 as a potential therapeutic target in diseases. These findings also expose the need for further investigations concerning the role of p62 in tumorigenesis, neurodegenerative diseases, and lipoprotein metabolism given that different gene mutations, cell types, and environmental conditions are known to dramatically influence its effects.
p62 plays a significant role in the regulation of selective autophagy via its interaction domains. However, little is known about the interaction domains of p62, which regulates selective autophagy, especially the ZZ domain, NLS domain, NES domain, and PEST. Moreover, autophosphorylation of p62 besides S403, have other phosphorylation sites to regulate protein aggregate formation. Furthermore, p62 plays a critical role in an oxidative stress response pathway via its direct interactions with the ubiquitin ligase adaptor Keap1.This interaction results in constitutive activation of the transcription factor NF-E2-related factor 2 (Nrf2) in neurodegenerative diseases and cancer (Nguyen et al., 2009). Whether the p62 interaction with keap1 relies on the KIR domain only or may involve other domains as well is unknown. Future studies on selective autophagy should address these questions.
Footnotes
Acknowledgments
This subject was supported by the Innovative Research Team for Science and Technology in Higher Educational Institutions of Hunan Province and Natural Science Foundation of China (No. 81070221) and Visiting Scholar Foundation of Key Laboratory for Biorheological Science and Technology (Chongqing University) of Ministry of Education (2010).
Disclosure Statement
No competing financial interests exist.