
Abstract
The seleno-organic compound and radical scavenger ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) have been extensively employed as an anti-inflammatory and neuroprotective compound. However, its glutathione peroxidase activity at the expense of cellular thiols groups could underlie certain deleterious actions of the compound on cell physiology. In this study, we have analyzed the effect of ebselen on rat hippocampal astrocytes in culture. Cellular viability, the intracellular free-Ca2+ concentration ([Ca2+]c), the mitochondrial free-Ca2+ concentration ([Ca2+]m), and mitochondrial membrane potential (ψm) were analyzed. The caspase-3 activity was also assayed. Our results show that cell viability was reduced by treatment of cells with ebselen, depending on the concentration employed. In the presence of ebselen, we observed an initial transient increase in [Ca2+]c that was then followed by a progressive increase to an elevated plateau. We also observed a transient increase in [Ca2+]m in the presence of ebselen that returned toward a value over the prestimulation level. The compound induced depolarization of ψm and altered the permeability of the mitochondrial membrane. Additionally, a disruption of the mitochondrial network was observed. Finally, we did not detect changes in caspase-3 activation in response to ebselen treatment. Collectively, these data support the likelihood of ebselen, depending on the concentration employed, reduces viability of rat hippocampal astrocytes via its action on the mitochondrial activity. These may be early effects that do not involve caspase-3 activation. We conclude that, depending on the concentration used, ebselen might exert deleterious actions on astrocyte physiology that could compromise cell function.
Introduction
The seleno-organic compound 2-phenyl-1,2-benzisoselenazol-3(2H)-one (ebselen) is a substance with the radical-scavenging activity (Fujisawa and Kadoma, 2005). The ebselen main effect involves attenuation of a free radical generation and detoxification of free radicals and the toxic products of their reactions. Its beneficial effects are mediated through its antioxidant and glutathione peroxidase-mimetic properties. Therefore, it has been extensively employed as an anti-inflammatory and neuroprotective compound.
Ebselen acts as an antiaging agent by attenuating oxidative stress in Caenorhabditis elegans (Avila et al., 2012), protects neurons from ischemic damage via control of the expressions of GABA shunt enzymes (Seo et al., 2009), has a marked inhibitory effect on neuronal damage during stroke (Yamagata et al., 2008), protects against excitotoxicity induced by glutamate in isolated chick retina (Centurião et al., 2005), and prevents ischemia-induced cytotoxicity and apoptosis (Gabryel and Małecki, 2006) and reactive oxygen species (ROS) production (Pérez-Ortiz et al., 2004) in astrocytes.
Epidemiological evidence suggests that overexposure to selenium can facilitate the appearance of chronic degenerative diseases. Under the ebselen's activity, as an enzyme mimic, the reaction catalyzed is that of a glutathione peroxidase. Substrates for the reaction vary from hydrogen peroxide and smaller organic hydroperoxides to membrane-bound phospholipids and cholesterol hydroperoxides, in addition to other suitable thiol compounds (Sies, 1993). Many cellular proteins contain cysteine residues, are central to cellular homeostasis, and are sensitive to redox status (Murphy, 2012a). Thus, agents that block protein surface thiols can disrupt the activity of enzymes and transporters, by means of changing their redox status (Antony and Bayse, 2011). Indeed, it has been found that concentrations of 20, 50, and 100 μM ebselen induce cell death (Yang et al., 2000; Brown et al., 2009), for which the mechanism remains unclear.
We have previously shown that ebselen induced Ca2+ release from the endoplasmic reticulum (ER). In addition, other signs were observed; those signs could be the basis of astroglial reactivity, which might evolve into impairment of astroglial physiology (Salazar et al., 2008). In spite of the great number of investigations carried out to demonstrate the actions of ebselen against established pathological events, the hypothesis that ebselen might exert side effects on normal cell physiology requires further attention.
Here we show that ebselen, beyond its putative protective role against harmful radicals, depicts a dark side on astroglial physiology. Ebselen could significantly compromise astroglial function, in such a way that it might lead to detrimental consequences in healthy cells.
Materials and Methods
Subjects
Newborn (24 h) Wistar rats were used for this study. Animals were obtained from the animal house of the University of Extremadura. Animal procedures were approved by the institutional Bioethics Committee.
Materials
Cell culture reagents, the Dulbecco's modified Eagle's medium (DMEM), Hank's balanced salts (HBSS), and Trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA) were obtained from Gibco BRL (Invitrogen, Barcelona, Spain). MITO+ (concentrated serum extender) and rat-tail collagen were obtained from BD Biosciences (Bedford, MA). Fetal bovine serum (FBS) was purchased from HyClone (Thermo Scientific, Erembodegen, Belgium). Penicillin/streptomycin was obtained from BioWhittaker (Lonza, Basel, Switzerland). Papain was obtained from Worthington Biochemical Corp. (Lakewood, NJ). AlamarBlue® was purchased from AbD serotec (bioNova Científica, Madrid, Spain). Ebselen, 1,2-Bis(2-amino-5-methylphenoxy) ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl) ester (Dimethyl BAPTA), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), N-acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (ACDEVD-AMC), and thapsigargin were obtained from Sigma Chemicals Co. (Madrid, Spain). Fura-2 acetoxylmethyl ester (fura-2/AM), MitoTracker™ Green FM, rhod-2 acetoxylmethyl ester (rhod-2/AM), tetramethylrhodamine methyl ester perchlorate (TMRM), and trypsin-EDTA were purchased from Invitrogen (Barcelona, Spain). All other reagents were of analytical grade.
Preparation of cell cultures
Subcultures of astrocytes were prepared following previously established methods (González et al., 2006). Briefly, following extraction of the brain, the hippocampus was dissected and subjected to digestion with papain during 1 h at 37°C. After enzymatic digestion the tissue was washed, resuspended in a culture medium consisting of DMEM supplemented with FBS (10%), penicillin (20,000 IU), streptomycin (0.1 mg/mL), and MITO+ (0.1%), and mechanically disgregated by gently pipetting the tissue through tips of small diameter. Cells were then plated in 75-cm2 flasks and allowed to grow in a culture medium at 37°C in a humidified incubator (5% CO2); cultures were confluent 10–14 days after seeding.
Thereafter, the culture medium was removed and replaced by the HBSS medium containing trypsin-EDTA. Cells were detached by 5-min incubation in this medium. The trypsinization step was stopped by adding a medium consisting of the culture medium supplemented with 0.25% bovine serum albumin and 0.25% trypsin inhibitor. Cells were centrifuged (50–60 g, 5 min) and resuspended in the culture medium. Finally, the cells were reseeded again in 75-cm2 flasks and grown in the culture medium in a humidified incubator at 37°C and 5% CO2. Cells reached confluence 10–12 days after reseeding. On the day of use, cells were detached by trypsinization, centrifuged (50–60 g, 5 min), and resuspended in a Na- N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid (HEPES) buffer containing (in mM) 140 NaCl, 4.7 KCl, 1.3 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, and 0.2% bovine serum albumin (pH adjusted to 7.4). The purity of astrocytes culture obtained with this technique has been previously assessed by GFAP immunostaining in our laboratory. In our conditions, we can assume that over 95% of the cells were astrocytes (González et al., 2006).
Cell viability assay
Analysis of cell proliferation and cytotoxicity under the different treatments applied was analyzed using AlamarBlue. This is a proven cell viability indicator that uses the natural reducing power of living cells. The active ingredient of AlamarBlue (resazurin) is a nontoxic, cell permeable compound that is blue in color and virtually nonfluorescent. Upon entering cells, resazurin is reduced to resorufin, which produces very bright red fluorescence. Viable cells continuously convert resazurin to resorufin, thereby generating a quantitative measure of viability and cytotoxicity.
To evaluate cell survival, once isolated, cells were centrifuged and transferred to a culture medium of the following composition: DMEM supplemented with FBS (10%), penicillin (20,000 IU), streptomycin (0.1 mg/mL), and MITO+ (0.1%). This medium was prepared under sterile conditions. Then, the cells were seeded onto independent Petri dishes (35-mm diameter) at a cell density of 30,000 per dish and incubated in the culture medium in a humidified incubator at 37°C and 5% CO2. Twenty-four hours after isolation, cells were washed twice with standard phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4; pH adjusted to 7.4) to remove death cells, and a fresh culture medium was added to the dishes. At this point, AlamarBlue was added to the dishes at the desired concentration and the cells were challenged with the stimuli. Determinations of cell viability were performed according to the manufacturer's directions.
Data of cell viability show the mean reduction of AlamarBlue expressed in percentage±S.E.M. (n) with respect to control (nonstimulated) cells, where n is the number of independent experiments.
Determination of intracellular free-Ca2+ concentration
Changes in [Ca2+]c were monitored following changes in fura-2-derived fluorescence. For dye loading of the cells, the culture medium was replaced by a physiological solution containing 130 mM NaCl, 4.7 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 1.2 mM KH2PO4, 10 mM glucose, 10 mM HEPES, and 0.2% bovine serum albumin (pH=7.4 adjusted with NaOH). Cultured cells were then loaded with the fluorescent ratiometric Ca2+ indicator fura-2 by incubation with fura-2/AM (4 μM) at room temperature (23°C–25°C) for 40 min as previously described (González et al., 2002). Changes in fura-2-derived fluorescence closely report changes in [Ca2+]c (Grynkiewicz et al., 1985).
For monitorization of Ca2+-dependent fluorescence signals, the coverslip with cultured astrocytes was mounted on an experimental perfusion chamber and placed on the stage of an epifluorescence inverted microscope (Nikon Diaphot T200, Melville, NY). The cells were continuously superfused with a control Na-HEPES buffer containing (in mM) 140 NaCl, 4.7 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose (pH adjusted to 7.4).
For fura-2 fluorescence changes determination, an image acquisition and analysis system for video microscopy was employed (Hamamatsu Photonics, Hamamatsu, Japan). Cells were alternatively excited with light from a xenon arc lamp passed through a high-speed monochromator (Polychrome IV, Photonics, Hamamatsu, Japan) at 340/380 nm. Fluorescence emission at 505 nm was detected using a cooled digital CCD camera (Hisca CCD C-6790, Hamamatsu, Japan) and recorded using dedicated software (Aquacosmos 2.5, Hamamatsu Photonics, Hamamatsu, Japan). All fluorescence measurements were made from areas considered individual cells.
All stimuli were dissolved in the extracellular Na-HEPES buffer, and applied directly to the cells in the perfusion chamber. When Ca2+ free conditions were applied, the Na-HEPES buffer contained no added Ca2+ and was supplemented with 0.5 mM EGTA. In a set of experiments, cells were loaded with dimethyl 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra acetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) (10 μM for 30 min), and then challenged with stimuli in the absence of extracellular Ca2+ (medium supplemented with 0.5 mM EGTA).
Experiments were performed at room temperature (23°C–25°C) and results are expressed as the mean ratio of fluorescence emitted at both excitation wavelengths previously normalized to the basal (resting) fluorescence±SEM, n (where n is the number of independent experiments/number of analyzed cells of each treatment).
Determination of mitochondrial free-Ca2+ concentration
Determining changes in mitochondrial Ca2+ signals ([Ca2+]m) was performed following previously established methods (González et al., 2000). Briefly, cells were loaded with rhod-2/AM (8 μM) at 4°C for 15 min. After incubation with rhod-2/AM, the extracellular medium was replaced by a fresh Na-HEPES buffer, and cells were incubated at room temperature (23°C–25°C) for 30 min. At the concentration used, rhod-2 accumulates within mitochondria. An increase in rhod-2 fluorescence reflects an increase in [Ca2+]m. For monitoring fluorescence signals, coverslips with dye-loaded cells were placed in a perfusion chamber on the stage of the above-mentioned confocal system. Using a×60 oil immersion objective fluorescence, images of 256×256 pixels with a resolution of 0.287 μm/pixel were recorded every 4 s. Excitation light at 543 nm from a 25-mW helium–neon laser was employed. Emitted fluorescence was collected through a 605/32-nm band-pass filter, employing different photomultipliers as previously described (González et al., 2003). Neutral density filters had to be employed to reduce laser intensity to 1%–3% and diminish photobleaching. Results are expressed as the absolute values of fluorescence emission at the excitation wavelength employed. Data were normalized previously to the basal (resting) fluorescence values.
Mitochondrial membrane potential determination
Changes in ψm were recorded using the dye TMRM as described previously (González et al., 2003). Cells were incubated during 30 min in the presence of 50 nM TMRM at 37°C. At the concentration used in our conditions, TMRM accumulates within mitochondria driven by the membrane potential, but autoquenching is negligible. A decrease in TMRM fluorescence reflects depolarization of ψm, because of diffusion of the dye to the cytosol. No TMRM was added to the medium after the initial loading period.
Fluorescence images from cells loaded with TMRM were obtained using the laser scanning confocal microscope mentioned earlier. Using a ×60 oil-immersion objective, fluorescence images of 256×256 pixels with a resolution of 0.287 μm/pixel were recorded every 4 s. TMRM was excited by the 543-nm line from a 25-mW helium–neon laser, and emission was collected through a 605/32-nm band-pass filter. Neutral density filters had to be employed to reduce laser intensity to 1%–3% and diminish photobleaching. Results are expressed as the absolute values of fluorescence emission at the excitation wavelength employed. Data were normalized previously to the basal (resting) fluorescence values.
Localization of mitochondria
Localization of mitochondria was assayed by incubation of astrocytes in the presence of MitoTracker Green FM. Loading of cells with MitoTracker Green FM (100 nM) was performed at room temperature (23°C–25°C) for 30 min. After incubation, the extracellular medium was replaced by the fresh Na-HEPES buffer. This dye has been employed as a mitochondrial marker (González et al., 2003).
For monitoring fluorescence signals, coverslips with dye-loaded cells were placed in a perfusion chamber on the stage of a confocal Nikon Eclipse TE300 microscope (Nikon Instruments, Inc.) and continuously superfused with the Na–HEPES buffer. Fluorescence images from cells loaded with Mito Tracker Green FM were obtained employing a confocal laser scanning system Bio-Rad MRC 1024 (American Laser Corp., Salt Lake City, UT). Using a ×60 oil immersion objective fluorescence, images of 256×256 pixels with a resolution of 0.287 μm/pixel were recorded every 4 s.
Excitation light at 488 nm from a 100-mW argon laser was employed. Emitted fluorescence was collected through a 522/35-nm band-pass filter employing a photomultiplier, as previously described (González et al., 2000). Laser intensity was reduced to 1%–3% with neutral density filters to reduce photobleaching. The software used for the imaging was Laser Sharp MRC-1024 Version 3.2 (Bio-Rad, Deisenhofen, Germany).
Caspase activity assay
To determine the caspase-3 activity, stimulated or resting cells were sonicated and cell lysates were incubated with 2 mL of a substrate solution containing 20 mM HEPES, pH 7.4, 2 mM EDTA, 0.1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), 5 mM dithiothreitol, and 8.25 μM of caspase-3 substrate, for 2 h at 37°C.
Substrate cleavage was measured at an excitation wavelength of 360 nm and emission at 460 nm. An ELISA spectrofluorimeter (Tecan Infinite M200, Grödig, Austria) was employed for the determinations. The activity of caspase was calculated from the cleavage of the specific fluorogenic substrate (AC-DEVDAMC). The data were calculated as fluorescence units per milligram of protein, and were presented as percentage±S.E.M. (n) with respect to control (nonstimulated) cells, where n is the number of independent experiments.
Statistical analysis
Statistical analysis of data was performed by one-way analysis of variance followed by the Tukey post hoc test, and only p-values<0.05 were considered statistically significant. For individual comparisons and statistics between individual treatments, we employed the Student's t-test and only p-values<0.05 were considered statistically significant.
Results
Effect of ebselen on cell viability
In a former work, we proposed that astroglial physiology could be altered by ebselen (Salazar et al., 2008). To investigate whether the antioxidant has any affect on cell viability, different batches of astrocytes were incubated for 24–96 h in the presence of no stimulus (control cells), ebselen (1, 10, and 100 μM), or 100 μM H2O2. Viability of astrocytes did not change significantly along the incubation period in the presence of 1 μM ebselen compared to control (unstimulated) cells, considered 100% (Fig. 1A). However, cell survival was reduced following incubation of cells in the presence of 10 and 100 μM ebselen, compared to control cells. The reduction of cell survival was dependent on the time of incubation and on the concentration of ebselen used (Fig. 1A). As a control, we incubated astrocytes in the presence of 100 μM H2O2, an oxidant with negative effects on cell viability. Under this treatment, cell survival also dropped with the time of incubation (Fig. 1B). Throughout the following experiments, we used 100 μM ebselen, because it exhibited the highest effect on cell viability.
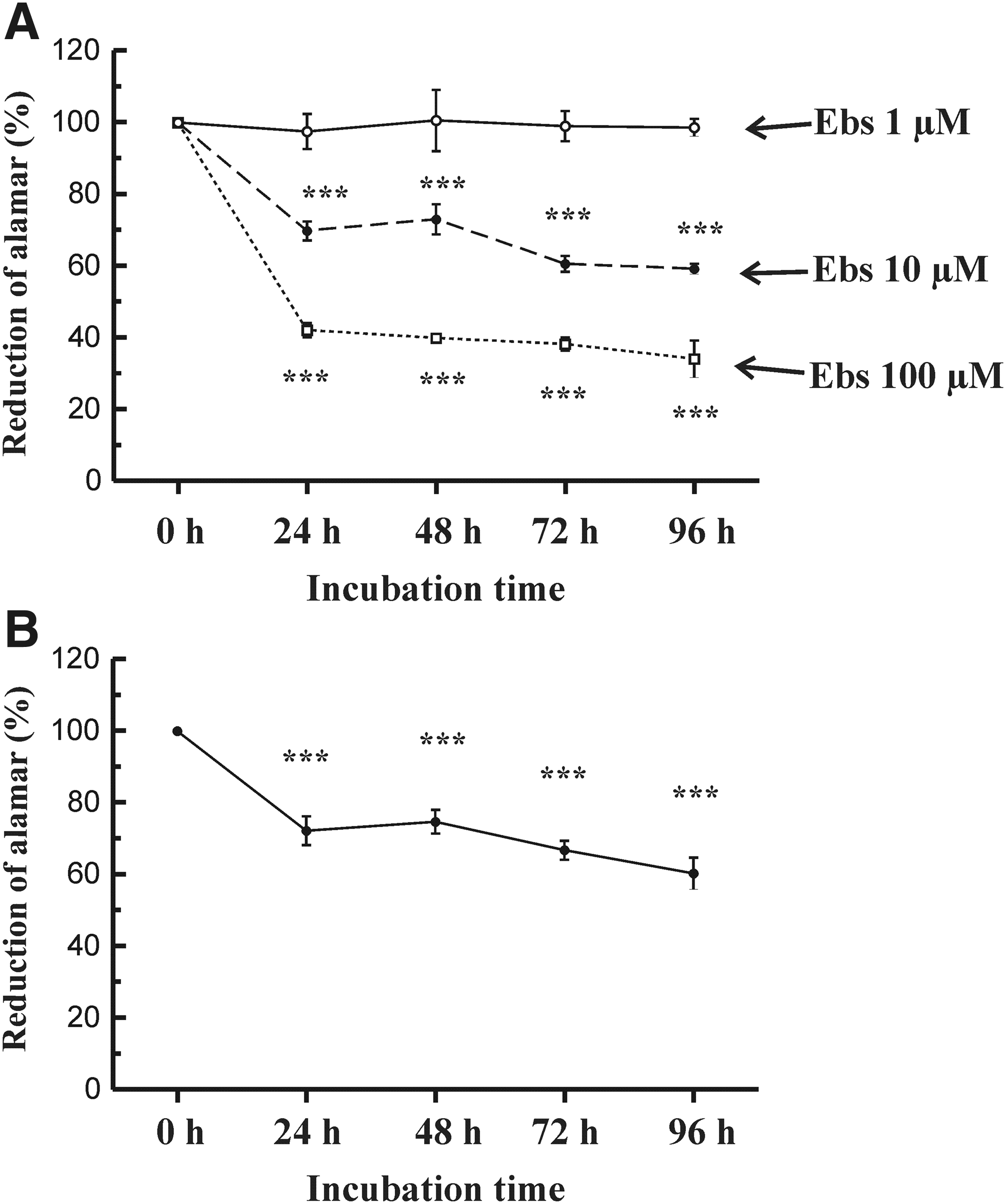
Analysis of viability of rat hippocampal astrocytes. Cell proliferation and cytotoxicity under the different treatments applied were analyzed studying AlamarBlue® reduction, as described in Materials and Methods.
Effect of ebselen on [Ca2+]c
Perfusion of astrocytes with ebselen (100 μM), in the presence of Ca2+ in the extracellular medium, led to an increase in [Ca2+]c, consisting of an initial transient increase that was then followed by a progressive increase to an elevated value over the prestimulation level (152 cells of 152 total cells studied, n=4 experiments; Fig. 2A).
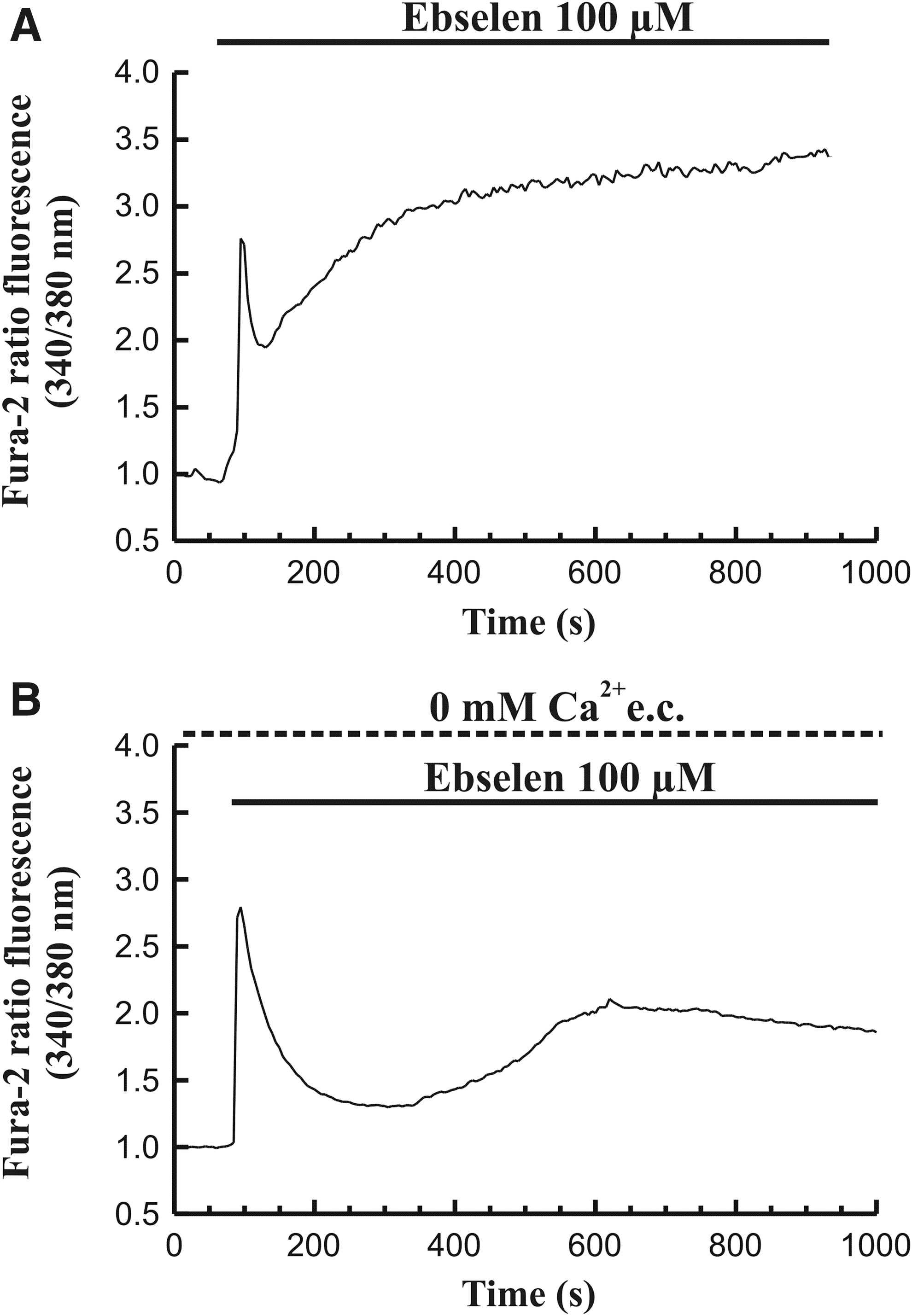
Monitorization of the effect of ebselen on [Ca2+]c.
On the other hand, stimulation of astrocytes with ebselen (100 μM) in the absence of Ca2+ in the extracellular medium (medium containing 0.5 mM EGTA) induced a transient increase in [Ca2+]c that was later followed by a slow and progressive increase toward a plateau over the prestimulation level (208 cells of 208 total cells studied, n=8 experiments; Fig. 2B). The plateau value of [Ca2+]c reached under these conditions was lower compared to that observed in the presence of extracellular Ca2+. These results reflect a release of the ion from intracellular stores.
A phenomenon characterized by appearance of irregular bulges, blebbing, at the plasma membrane was observed following the increase in [Ca2+]c, when the cells were stimulated with ebselen (100 μM) in the presence of Ca2+ in the extracellular medium. When astrocytes were stimulated with the antioxidant in the absence of extracellular Ca2+ (medium containing 0.5 mM EGTA), membrane blebbing was delayed in time, compared to the experiments performed in the presence of extracellular Ca2+. Similar results were obtained when the cells were challenged with ebselen (100 μM) in the additional presence of 10 μM of the intracellular Ca2+ chelator dimethyl-BAPTA (data not shown).
Effect of ebselen on mitochondria activity
In our experiments, we employed different fluorescent dyes that selectively accumulate into mitochondria, and report changes in physiological parameters within these organelles. Mitochondria are organelles with a high ability to sequester Ca2+ when it is released from the ER. Accumulated Ca2+ into mitochondria could mean a stimulatory signal that serves to modify the organelle activity (González et al., 2000; González et al., 2003). Ca2+ released by ebselen could then accumulate into mitochondria. As expected, when rhod-2-loaded cells were stimulated with 100 μM ebselen, we observed a transient increase in fluorescence, related to increase in [Ca2+]m. Thereafter, [Ca2+]m returned toward a plateau value over the prestimulation level (n=4 exp/18 cell/90 mitochondrial areas) (Fig. 3).

Effect of ebselen on [Ca2+]m in rat hippocampal astrocytes. Time course of changes in [Ca2+]m in response to 100 μM ebselen. A horizontal bar shows the time during which the stimulus was applied to the cells. The graphs are representative of four independent experiments. [Ca2+]m, mitochondrial free-Ca2+ concentration.
On the other hand, the mitochondrial electron transport chain provides the energy that drives the diffusion of protons into the matrix promoting ATP synthesis, and the function of mitochondria depends on the maintenance of a ψm. In relation to a change in [Ca2+]c, depolarization of ψm occurs, which depends on Ca2+ uptake by mitochondria (González et al., 2000; González et al., 2003). Thus, it could be possible that ebselen had an effect on ψm. Therefore, we performed a series of experiments in which astrocytes were loaded with the mitochondrion-specific voltage-sensitive dye TMRM. As expected, when cells were perfused with 100 μM ebselen which, as we have shown, induces an increase in both [Ca2+]c and [Ca2+]m, it could be observed a depolarization of ψm (n=5 exp/38 cell/190 mitochondrial areas). Under our experimental conditions, there was no recovery of ψm (Fig. 4).
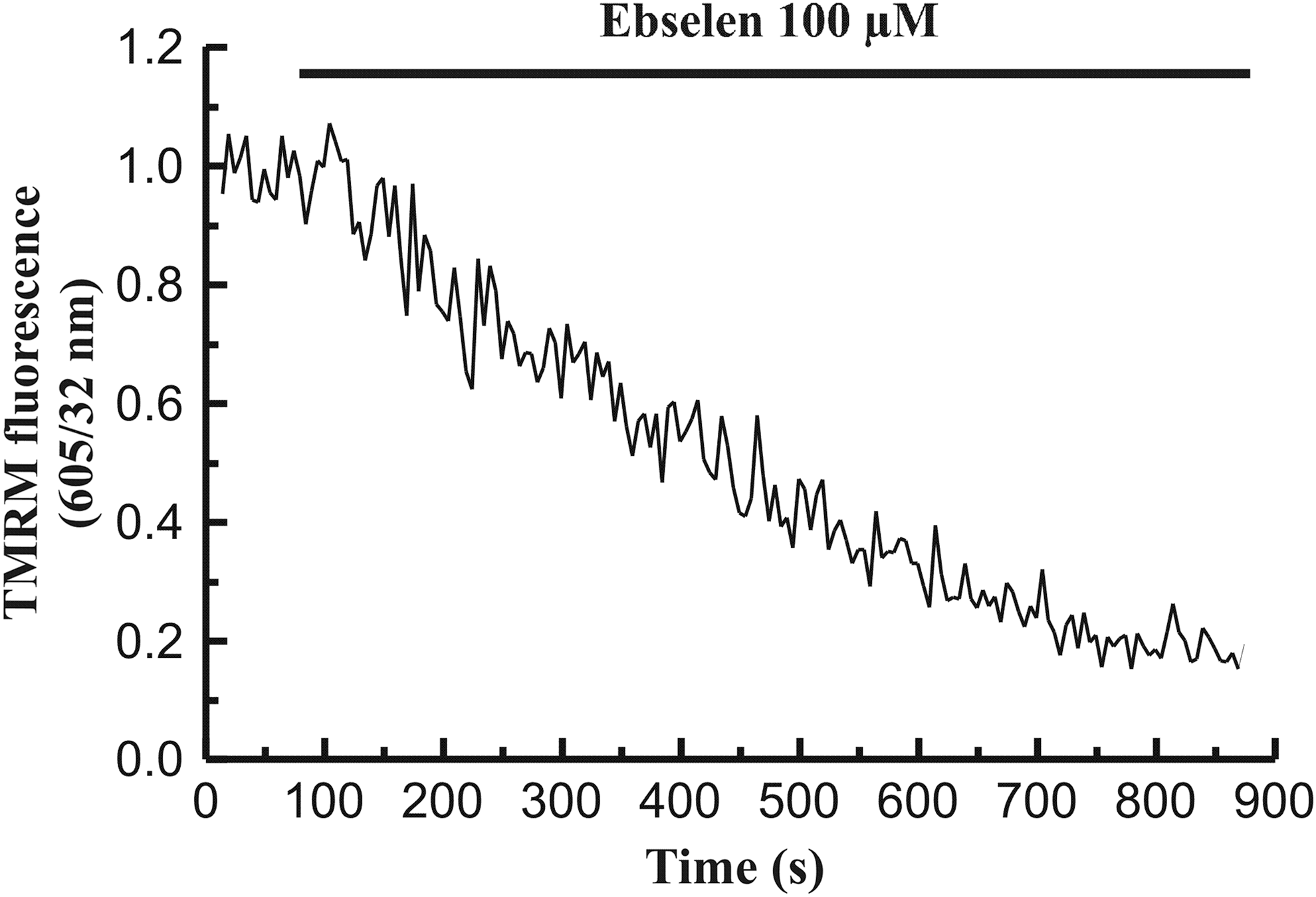
Effect of ebselen on mitochondrial membrane potential (ψm) in rat hippocampal astrocytes. Time course of changes in ψm in response to 100 μM ebselen. A horizontal bar shows the time during which the stimulus was applied to the cells. The graphs are representative of six independent experiments.
The described effects for treatment of cells with 100 μM ebselen on [Ca2+]m and ψm, were also observed when astrocytes were incubated in the presence of 20 or 50 μM ebselen (data not shown).
Because prolonged depolarization of mitochondria could be the basis of a mitochondrial stress that could interfere with its functional network, we were also interested in evaluating the distribution of mitochondria within the cells. Localization of mitochondria was assayed by incubation of cells in the presence of MitoTracker Green FM, a dye that selectively accumulates inside these organelles (González et al., 2003). Figure 5A shows a representative fluorescence image of control (unstimulated) astrocytes labeled with MitoTracker Green FM. We could observe a consistent network of mitochondria, which appeared distributed all through the cytosol. Following 5 min-incubation of astrocytes in the presence of 100 μM ebselen, a disruption of the mitochondrial network was observed (Fig. 5B).
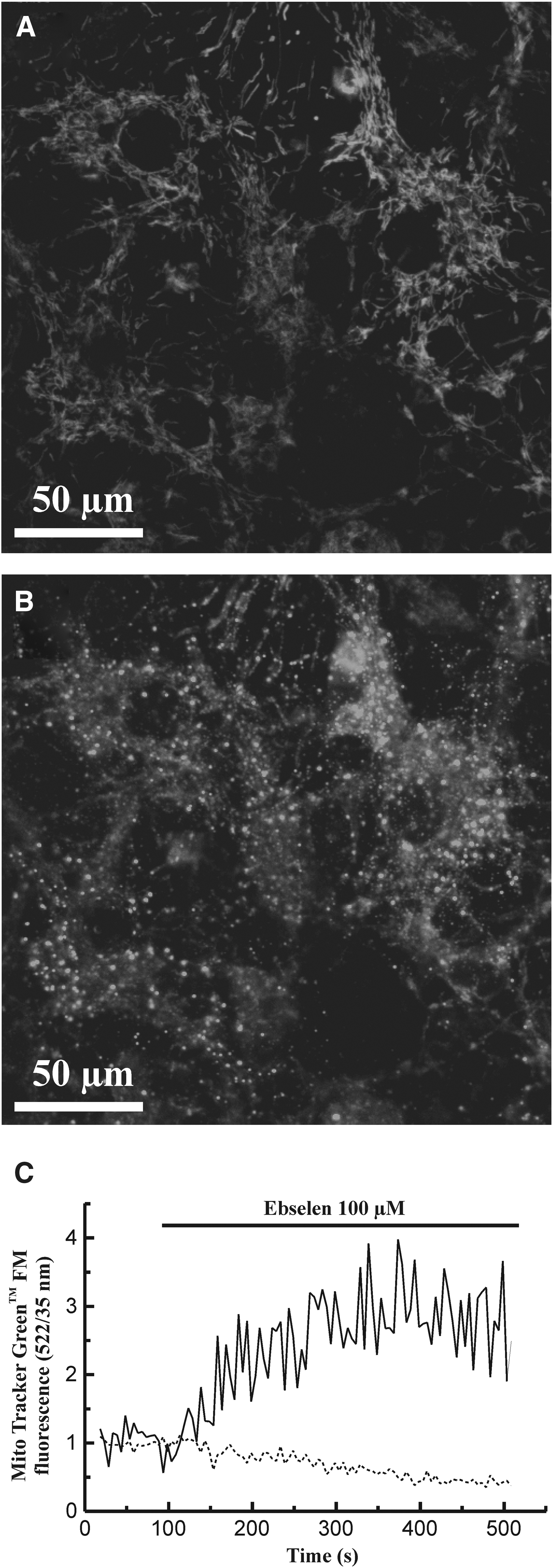
Confocal microscopy studies of rat hippocampal astrocytes loaded with the fluorescent probe Mito Tracker™ Green FM. Employing confocal laser scanning microscopy, bright fluorescent spots could be detected, being spread all through the cytosolic area.
In addition, we observed an increase of MitoTracker Green FM-derived fluorescence in the cytosol, following treatment of cells with ebselen (Fig. 5C, continuous line). This was accompanied by a decrease in mitochondrial fluorescence (Fig. 5C, dashed line). This reflects an increase in mitochondrial membrane permeability that leads to appearance of the dye, which was initially trapped within mitochondria, in the cytosol (n=5 exp/17 cell).
Caspase activity assay
The effect of ebselen that we have observed could be a starting point for the process of apoptosis. Therefore, we examined the effect of the antioxidant on caspase-3 activation. For this purpose, rat hippocampal astrocytes were incubated during 15 min in the presence of 100 μM ebselen. Under these conditions, we did not observe significant changes in caspase-3 activation (1.45%±0.98%) compared to unstimulated cells. As a control, treatment of cells with 100 μM H2O2, a compound that promotes apoptosis (Kaufmann et al., 2003), increased the caspase-3 activity by a 20.50%±2.25% (n=5; p<0.001).
Discussion
Therapy with antioxidants has been extensively used in the treatment of illness in different tissues, including the brain. In this line, the seleno-organic compound ebselen, which was originally developed as an anti-inflammatory agent, has been shown to protect against damage caused by free radicals in the CNS (Pérez-Ortiz et al., 2004; Green and Ashwood, 2005; Funchal et al., 2006; Gabryel and Małecki, 2006; Ghisleni et al., 2008). However, in spite of the positive effects of ebselen, certain signs were observed in response to selenium-containing compounds that could be the basis of potential damaging actions. In this respect, recent points of epidemiological evidence suggest that overexposure to selenium can facilitate the appearance of chronic degenerative diseases. However, the molecular mechanisms are not fully understood.
It has been shown that selenium-containing compounds are able to open Ca2+ release channels at the sarco ER membrane, leading to an increase in [Ca2+]c (Xia et al., 2004). Moreover, emptying of the Ca2+ content in the ER has been related to cell stress and impairment of cell physiology (Fradejas et al., 2010; Kim et al., 2010). In agreement to this, we have shown in a previous work that ebselen induced Ca2+ release from the ER (Salazar et al., 2008).
On the other hand, a leading mechanism of selenium cytotoxicity is a change in the oxidative state of cellular systems, with a resultant production of ROS (Wallenberg et al., 2010).
Mitochondria are organelles with a major function in the cellular oxidative metabolism. The mitochondrial matrix contains many of the central processes of metabolism (Duchen, 2004), depicting Ca2+ an important role in mitochondrial physiology (Duchen et al., 2008).
Ebselen mimics glutathione peroxidase, changing therefore the cell redox status (Antony and Bayse, 2011). Selenium can also interact with endogenous -SH groups (Nogueira and Rocha, 2011). Moreover, a range of mitochondrial proteins containing cysteine residues is central to mitochondrial homeostasis, and is sensitive to redox status. And agents that block protein surface thiols can disrupt the activity of enzymes and transporters (Murphy, 2012b). Derived mitochondrial dysfunction has been implicated in cellular disorders (Dröse and Brandt, 2012).
Concentrations of ebselen, ranging from 1 to 40 μM, have been widely employed in actual and past studies on different tissues and cell types, where it shows a protective action (Yamagata et al., 2008; Deng et al., 2010; Hassan et al., 2010; Yue et al., 2011; Du et al., 2012). However, as stated in the introduction, ebselen might induce cell death at certain concentrations.
Here we show that ebselen induces Ca2+ mobilization from intracellular stores and leads to an elevated [Ca2+]c, confirming our previous observations (Salazar et al., 2008). The effect of ebselen on [Ca2+]c could not be inhibited by incubation of astrocytes in the absence of extracellular Ca2+, or by incubation of cells in the presence of the intracellular Ca2+ chelator dimethyl-BAPTA.
Taking into account the above-mentioned effects of selenium, we hypothesized that the Ca2+-mobilizing action of ebselen could be injurious to the cell, and we investigated its effects on several mitochondrial parameters.
Our results show a transient accumulation of Ca2+ into mitochondria, following treatment of cells with ebselen. Thus, ebselen treatment might induce changes in mitochondrial function.
In addition to [Ca2+]m, in our study, we have also noticed changes in other mitochondrial parameters in the presence of ebselen. First, we have observed a depolarization of ψm.
In a former work, it has been shown that selenium can potentially influence the activity and function of oxidoreductases within the cell (Wallenberg et al., 2010). These proteins carry out a central function in maintaining the redox balance within the cell, and their imbalance can contribute to selenium toxicity. Indeed, a recent work has shown that ebselen caused a statistically significant inhibition of the mitochondrial complexes I and II activity, an effect completely reversed by reduced glutathione (Puntel et al., 2013). The authors suggest that ebselen-induced mitochondrial complexes I and II inhibition can be mediated by a thiol oxidation activity, that is, ebselen can oxidize critical thiol groups from mitochondrial complexes I and II. As a consequence, inhibition of mitochondrial respiratory chain complexes will lead to mitochondrial depolarization, as we have observed.
Second, we have also observed a disruption of mitochondrial network. Mitochondria form a dynamic network responsible for energy production, Ca2+ homeostasis, and cell signaling. Appropriate distribution of the mitochondrial network contributes to organelle function and is essential for cell survival (Frederick and Shaw, 2007). Additionally, redistribution of mitochondria within the cell can be a component of regulatory pathways (Murphy, 2012b). It has been proposed that fragmentation of the mitochondrial network is related neurodegeneration (Almajan et al., 2012). In agreement to these observations, the effect of ebselen on mitochondrial network could be deleterious to astrocyte physiology.
And third, ebselen induced an increase in mitochondrial membrane permeability. A common process associated with oxidative stress and severe mitochondrial impairment is the increase in mitochondrial membrane permeability, as described in many neurodegenerative diseases (Eckmann et al., 2013). The consequences of such an alteration of mitochondrial physiology are not uncommon. As expected, ebselen induces a decrease in cell viability.
In a previous work, we have shown that ebselen (1–10 μM) did not alter the integrity of the cellular membrane of astrocytes, indicated as absence of lactate dehydrogenase release to the extracellular medium. However, in the presence of 20 μM ebselen, it was detectable a slight, but significant decrease of astrocyte viability (Salazar et al., 2008).
In the present study, we demonstrate that cell viability is decreased in the presence of ebselen, as reflected by the studies on the reducing power of living cells. The analysis employing the growth indicator AlamarBlue, are based on the detection of cellular metabolism. Its active compound resazurin is effectively reduced in mitochondria, making it useful also to assess the mitochondrial metabolic activity. Thus, in addition to its utility as a marker of diminished cell viability, the decreased reduction of AlamarBlue that we have observed is an index of decreased mitochondrial function. Therefore, our observations agree with a deleterious action of ebselen on cell survival, which might be a consequence of the evoked changes in mitochondrial physiology that we have observed.
On the other hand, we did not observe involvement of the caspase-3 activity in the actions of ebselen. In our hands, the effects of this compound could be early ones that might lead to an impairment of mitochondrial function that, in turn, could impair cellular homeostasis and induce cell damage without involving apoptosis.
In conclusion, our results confirm that ebselen induces changes in mitochondrial parameters that could compromise the function of the major energy synthesizing factory within the cell. If mitochondrial homeostasis is impaired, then astrocyte function and survival could be compromised. Causing mitochondrial dysfunction may be considered an important factor in the toxicity of ebselen.
Footnotes
Acknowledgments
Funding for this study was provided by Plan Regional de Investigación Sanitaria 2010 (PRIS10014) and Junta de Extremadura-FEDER (GR10010). Patricia Santofimia-Castaño was granted a fellowship from Junta de Extremadura and European Social Fund. The funding source(s) had no further role in the study design, in the collection, analysis, and interpretation of data, in the writing of the report, and in the decision to submit the article for publication. The authors would like to thank Mrs. Mercedes Gómez Blázquez for her valuable technical assistance.
Disclosure Statement
The authors declare that there is no conflict of interests.