
Abstract
Introduction
Numerous studies confirm that posttranscriptional regulations tightly control the clearance of ABCA1 mRNA as well as the trafficking, stability, degradation, and activity of the ABCA1 protein. MiR-33 targets the 3′-untranslated region (3′-UTR) of ABCA1 mRNA, which results in mRNA cleavage and reduction of protein generation (Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). ABCA1 palmitoylation accelerates subcellular trafficking of ABCA1 toward the plasma membrane, leading to elevation of cell-surface expression and lipid efflux ability of ABCA1 in COS cells (Singaraja et al., 2009). Our results show that the mimic peptide D4-F stabilizes the ABCA1 protein and increases ABCA1 levels in mouse macrophage cells (Liu et al., 2010). In addition, we have also identified that the unsaturated fatty acids oleate and eicosapentaenoic acid potently impair ABCA1 protein stability and facilitate ABCA1 clearance in THP-1 macrophages (Tang et al., 2003; MacDonald et al., 2009). Pharmacological inhibition of calpain-mediated ABCA1 degradation increases ABCA1 protein levels and lipid efflux function (Yokoyama et al., 2012). These results demonstrate the significance of posttranscriptional regulations in the modulation of ABCA1 protein expression and lipid metabolism.
Considering the critical role in ABCA1 expression, posttranscriptional mechanisms have been investigated by a number of recent studies. This review focuses on the recent progress of posttranscriptional regulations and the underlying mechanisms in ABCA1 expression and cholesterol efflux function. These pathways may provide a new avenue to heighten ABCA1 expression and reverse cholesterol transport (RCT).
The Clearance of ABCA1 mRNA
MicroRNAs comprise a class of small noncoding RNAs approximately 22 nucleotides in length and represent an elegant mechanism of posttranscriptional control of gene expression. Through this regulatory pathway, several microRNAs promote ABCA1 mRNA cleavage and decrease ABCA1 protein generation by partial complementary binding to 3′-UTR of ABCA1 mRNA (Rayner et al., 2012). A number of studies highlight the regulatory role of miR-33 in ABCA1 expression (Moore et al., 2010; Fernandez-Hernand et al., 2011). Other microRNAs, including miR-758, miR-10b, and miR-27, have also been discovered to induce ABCA1 mRNA degradation (Moore et al., 2011; Ramirez et al., 2011; Wang et al., 2012). (Table 1).
MicroRNAs partially complementarily bind to 3′-UTRs in ABCA1 mRNA and result in mRNA removal and translational repression. Thus, they inhibit intracellular cholesterol efflux in various cell/animal models.
miR-33
MiR-33 is an intronic microRNA with two mature forms. The miR-33 gene resides within the sterol response element-binding protein (SREBP) gene, and is concomitantly expressed and processed with the host gene (Brown et al., 2010; Horie et al., 2010; Moore et al., 2010; Fernandez-Hernand et al., 2011). In human, miR-33 has two isoforms: miR-33b, which is present in intron 17 of the SREBP-1 gene on chromosome 17; and miR-33a, which is located in intron 16 of the SREBP-2 gene on chromosome 22. In mice, there is only one miR-33 isoform that is conserved with human isoform miR-33a and located within intron 15 of the mouse SREBP-2 gene (Najafi-Shoushtari et al., 2010; Ingrid and Gelissen, 2010). MiR-33a and miR-33b differ by only two nucleotides outside the seed sequence (Gerin et al., 2010). The gene location and the sequences of the miR-33 family are highly conserved, which indicates the important regulatory role of miR-33 in vivo.
Two functional miR-33 responsive elements are possessed in human ABCA1 3′-UTR. A proximal element contains three overlapping putative miR-33-binding sites at nucleotides 120–172 downstream of the stop codon. A distal element with one putative miR-33-binding site is at nucleotides 1465–1481 after the stop codon. The same putative sequences are found in nucleotides 120–171 and 1426–1442 in the mouse ABCA1 gene. MiR-33 potently represses ABCA1 expression by binding to these binding sites within its mRNA 3′-UTR (Gerin et al., 2010; Moore et al., 2010; Rayner et al., 2010). MiR-33 overexpression strongly decreases the abundance of ABCA1 mRNA and protein in macrophages (Najafi-Shoushtari et al., 2010; Rayner et al., 2010) and mouse liver (Marquart et al., 2010; Fernandez-Hernando et al., 2013). Suppression of endogenous miR-33 profoundly increases ABCA1 levels in macrophage and hepatocyte cell lines (Horie et al., 2010, 2012; Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). When the miR-33 responsive elements are mutated, miR-33-dependent repression of ABCA1 expression is lost (Marquart et al., 2010). MiR-33 dramatically reduces ABCA1 protein levels, resulting in intracellular cholesterol accumulation (Moore et al., 2011; Rayner et al., 2011b; Sacco and Adeli, 2012; Wijesekara et al., 2012).
miR-758
MiR-758, a novel microRNA regulator, is identified as a second microRNA targeting ABCA1. MiR-758 is an intergenic microRNA and particularly abundant in the brain and liver of mouse tissues (Rayner et al., 2012). Although the human ABCA1 mRNA 3′-UTR has 2 computationally predicted miR-758-binding sites (site 1: 1501–1507; site 2: 3271–3277), miR-758 markedly represses ABCA1 expression through site 2 (3271–3277). MiR-758 transfection represses the expression of ABCA1 in human and mouse macrophages, and conversely, miR-758 inhibition increases ABCA1 expression. MiR-758 is more highly expressed than miR-33 in mouse brain, which suggests a potential, critical, regulatory role of miR-758 in ABCA1 expression in brain tissue (Ramirez et al., 2011).
miR-10b
MiR-10b was recently established to be involved in the posttranscriptional regulation of ABCA1 expression (Wang et al., 2012). MiR-10b directly interacts with the 3′-UTR of ABCA1 mRNA and decreases ABCA1 expression. Mutations in the seed sequence of the 3′-UTR of ABCA1 mRNA abrogate the miR-10b repression of ABCA1 expression. MiR-10b overexpression causes the appreciable reduction of ABCA1 expression and lipid removal in macrophage cells. MiR-10b levels are inversely related with ABCA1 expression levels in vivo. The inhibitory effect of miR-10b on ABCA1 expression has a long duration. These results confirm that miR-10b inhibits ABCA1 expression and macrophage cholesterol efflux (Wang et al., 2012).
miR-27
Our unpublished results indicate miR-27 as a critical regulator of ABCA1 by targeting ABCA1 mRNA. MiR-27 is predicted to bind to the 3′-UTR of ABCA1 mRNA using online databases. Human ABCA1 mRNA 3′-UTR contains two miR-27-binding sites (site 1: 2284–2290; site 2: 2592–2598). As anticipated, our experimental studies showed that miR-27 overexpression suppresses the expression of the ABCA1 protein in macrophage cells. Anti-miR-27 leads to the elevation of ABCA1 expression. These data suggest that miR-27 targets and degrades ABCA1 mRNA (Chen et al., 2012; Vickers et al., 2013).
Collectively, these microRNAs that target ABCA1 mRNA and regulate ABCA1 levels are promising/potential therapeutic targets for the prevention and treatment of dyslipidemia and atherosclerosis. For instance, anti-miR-33 leads to a sustained increase in plasma high density lipoprotein (HDL) and atherosclerotic plaque regression in nonhuman primates, which indicates the therapeutic potential of miR-33 inhibition in patients with atherosclerosis (Rayner et al., 2011a). Reduced miR-10b levels enhance macrophage cholesterol efflux and atherosclerosis regression in apoE−/− mouse, suggesting miR-10b as a potential target for human atherosclerotic disease (Wang et al., 2012). In vivo studies of miR-758 and miR-27 are sparse presently. MiR-758 may play an important role in neural lipid metabolism, and miR-27 in hepatic. Given the crucial roles of microRNAs in lipid metabolism and atherosclerotic disease, the manipulation of these microRNAs to raise ABCA1 levels is worth more detailed investigation (Pushparaj et al., 2008; Ouimet and Moore, 2013).
The Trafficking of ABCA1 Protein
Existing results have outlined the subcellular itinerary that traffics the intracellular ABCA1 protein to the cell surface (Kang et al., 2010). Nascent ABCA1 reaches the plasma membrane through anterograde vesicular transport (van Vliet et al., 2003). Newly synthesized ABCA1 undergoes proper folding and dimerization at the endoplasmic reticulum (ER). Dimerized ABCA1 is transported from the ER to the Golgi apparatus via vesicle transport. After palmitoylation in the Golgi, ABCA1 arrives at the trans-Golgi network (TGN). Then, ABCA1 is sorted for delivery to the plasma membrane or endosomes. After arriving at the plasma membrane, surface ABCA1 dynamically recycles between the plasma membrane and intracellular endocytic compartments (Neufeld et al., 2001; Kang et al., 2010). Plasma membrane-derived ABCA1 undergoes internalization by clathrin-mediated endocytosis. The internalized ABCA1 traffics to early endosomes, late endosomes, and recycling endosomes. The endocytosed ABCA1 partially returns back to the plasma membrane via recycling endosomes (Santamarina-Fojo et al., 2001; Neufeld et al., 2004). This subcellular trafficking pathway plays a key role in regulating cell-surface ABCA1 expression. (Fig. 1).

Schematic summary of the intracellular trafficking pathway of the ABCA1 protein. A newly synthesized ABCA1 protein undergoes N-glycosylation modification, proper folding, and homodimerization at ER. ABCA1 dimer is delivered to Golgi apparatus with the COPII-coated vesicle transport and the guidance of Rab1 and Rab2. The ABCA1 protein is palmitoylated by DHHC8 in the Golgi membrane. The xLxxKN motif directs palmitoylated ABCA1 to reach TGN. The ABCA1 protein is conveyed to the plasma membrane or early endosome via the TGN transport. Rab6, Rab8, and palmitoylation involve in the TGN to plasma membrane trafficking. Rab8, clathrin, and BIG1 implicate in the TGN to early endosome trafficking. Once arrived at the plasma membrane, ABCA1 undergoes internalization by clathrin-mediated endocytosis. Internalized ABCA1 dynamically traffics between the plasma membrane, recycling endosomes, early endosomes, and late endosomes. A number of Rabs and accessory proteins accelerate or suppress the shuttle of ABCA1 between the plasma membrane and intracellular vesicles. ER, endoplasmic reticulum; TGN, trans-Golgi network; BIG1, Brefeldin A-inhibited guanine nucleotide-exchange protein 1; CTSD, cathepsin D.
Recent studies revealed that accessory proteins and molecular structures indispensably are involved in the trafficking process of the ABCA1 protein. The palmitoyl transferase DHHC8 enzyme palmitoylates ABCA1 at cysteine residues 3, −23, −1110, and −1111 and promotes its localization at the plasma membrane (Singaraja et al., 2009). A highly conserved xLxxKN motif located within the N-terminal signal sequence of ABCA1 serves as a Golgi exit signal, directing the ABCA1 transporter to a post-Golgi vesicular sorting station and accelerating the vesicular transport of ABCA1 to the cell surface (Beers et al., 2011). Cathepsin D, a lysosomal protease, sorts ABCA1 from late endosomes and promotes subcellular trafficking of ABCA1 toward the plasma membrane in both macrophages and CHO cells (Haidar et al., 2006). Brefeldin A-inhibited guanine nucleotide-exchange protein 1 (BIG1) regulates vesicle trafficking between the TGN and the plasma membrane, and facilitates the ABCA1 protein recycling between the hepatocellular cell surface and endocytic compartments (Lin et al., 2013). Rab8 is involved in the trafficking of ABCA1, from the TGN to the plasma membrane and from endosome to the plasma membrane, facilitating ABCA1 cell surface expression in primary human macrophages (Linder et al., 2009; Jean and Kiger, 2012). Rab4A and Rab4B inhibit the recycling step from endosomes to the plasma membranes, and accelerate the endocytosis of ABCA1 from the plasma membrane to endocytic compartments (Tanaka et al., 2008). A deletion mutant of the proline-glutamate-serine-threonine (PEST) motif leads to the impaired internalization of ABCA1 and increased cholesterol efflux in HEK293 cells (Chen et al., 2005). These factors participate in the intracellular transport of ABCA1 and exert an influence on ABCA1 subcellular distribution.
Some factors interfere in the subcellular trafficking of ABCA1, resulting in the alteration of its localization at the plasma membrane. These factors are summarized as follows (Table 2).
These multiple factors impact ABCA1 protein trafficking, stability, degradation, and activity, respectively, in various cell/animal models. Cholesterol efflux is enhanced on the condition of increased vesicle transport toward plasma membrane, heightened ABCA1 protein stability, retarded ABCA1 protein degradation, and improved ABCA1 transport efficiency.
SPTLC1, serine palmitoyltransferase enzyme 1; OSBP, oxysterol-binding protein; CSN, COP9 signalosome.
Proteinic factors
A few proteinic factors influence in the subcellular trafficking of ABCA1. ApoA-I enhances the recycling of ABCA1 to the cell surface and increases cell surface ABCA1 (Lu et al., 2008; Zha et al., 2003). Pepstatin and prosaposin block the activity and expression of Cathepsin D, which cause the retention of ABCA1 in lysosomal compartments and reduces its trafficking to the plasma membrane (Haidar et al., 2006). The serine palmitoyltransferase enzyme 1 (SPTLC1) negatively regulates the ABCA1 ER exit, leading to ABCA1 retention at this organelle and reducing surface ABCA1 expression (Tamehiro et al., 2008; Kang et al., 2010). These proteinic factors affect ABCA1 distribution and function by targeting the processes of subcellular trafficking.
Compounds
Several compounds intervene in the subcellular trafficking of ABCA1, resulting in alterations to ABCA1 distribution. Myriocin pharmacologically inhibits SPTLC1, results in the disruption of the SPTLC1–ABCA1 complex and increases the levels of cell surface ABCA1 (Tamehiro et al., 2008). Brefeldin A and Monensin block the delivery of newly synthesized proteins to the cell surface, which causes accumulation of ABCA1 in the Golgi/ER, and in late endosomes and lysosomes (Neufeld et al., 2001). Cyclosporin A suppresses ABCA1 shuttle between the plasma membrane and intracellular endocytic compartments and traps the ABCA1 protein at the plasma membrane in mouse macrophages and BHK/ABCA1 cells (Le Goff et al., 2004; Nagao et al., 2013).
Since the subcellular trafficking and distribution of ABCA1 play critical roles in ABCA1-mediated cholesterol efflux and the subsequent RCT, the agonists should be urgently developed to facilitate the subcellular trafficking of the ABCA1 protein. For instance, myriocin-like agonists may promote ABCA1 palmitoylation and facilitate the trafficking of ABCA1 toward the cell surface. Thus, they may represent potential therapeutic targets for enhancing cholesterol efflux and inhibiting the atherosclerosis progress.
The Stability of ABCA1 Protein
Enhancing ABCA1 protein stability will increase the ABCA1 protein at the plasma membrane and in endosomes, and will impede subsequent intracellular degradation of the protein. Multiple factors have been recognized to modulate ABCA1 protein stability and alter its subcellular localization. (Table 2).
Helical apolipoproteins and mimic peptides
Helical apolipoproteins, including apoA-I, apoA-II, and apoE, as well as peptides such as L37pA, D37pA, and D4-F that are artificially synthesized to mimic amphiphilic helical segments of apolipoproteins stabilize ABCA1 through pathways discussed below.
First, helical proteins and mimic peptides activate protein kinase Cα (PKCα) or protein kinase A (PKA) signaling to phosphorylate serine and/or threonine residues in specific amino acid residues outside the PEST motif of ABCA1 to stabilize it (Martinez et al., 2003; Yamauchi et al., 2003; Liu et al., 2010; Mulay et al., 2012). The phosphorylation of these serine and/or threonine residues correlates with its stabilization (Arakawa et al., 2004; Buechler and Bauer, 2011). ApoA-I stabilizes total ABCA1 protein levels and increases its level at the cell membrane and in intracellular vesicular compartments (Mogilenko et al., 2012). This effect is suppressed by PKC inhibitors (Yamauchi et al., 2003). ApoA-II and apoE also stabilize the ABCA1 protein. Mimic peptides, L37pA and D37pA, which contain two class-A amphiphilic helices, can phosphorylate and stabilize the ABCA1 protein, resulting in higher levels of the ABCA1 protein in THP-1 and WI-38 cells (Arakawa et al., 2004). The mimic peptide D4-F, which contains one class-A amphiphilic helix, also enhances ABCA1 serine phosphorylation and ABCA1-dependent cholesterol efflux through the Cdc42/cAMP/PKA pathway in THP-1 macrophage-derived foam cells. Reduction of ABCA1 serine phosphorylation by siRNA targeting PKA decreases the levels of the ABCA1 protein (Liu et al., 2010). In addition, it has been reported that a site on the fourth extracellular loop is involved in regulating ABCA1 stability (Mukhamedova et al., 2007). Phosphorylation of serine and/or threonine residues within the fourth extracellular loop is closely associated with the improvement of ABCA1 protein stability. It is rational to speculate that the phosphorylation sites by the PKCα pathway may overlap the fourth extracellular loop of ABCA1.
Second, the residues, Thr-1286 and Thr-1305, in the PEST motif within the first intracellular loop of ABCA1 are constitutively phosphorylated. Helical apolipoproteins and mimic peptides can promote the PEST motif dephosphorylation, which leads to the stabilization of the ABCA1 protein residing on the plasma membrane (Martinez et al., 2003). For instance, free apoA-I increases surface ABCA1 protein stability in parallel with the dephosphorylation of the Thr residues in the ABCA1 PEST motif. The replacement of T1286 and T1305 with Ala impairs ABCA1 dephosphorylation and blocks PEST-dependent ABCA1 stabilization upon helical apolipoproteins and mimic peptides treatment (Martinez et al., 2003; Tang et al., 2004; Lee and Parks, 2005; Yokoyama, 2006; Lu et al., 2008). These results indicate that the dephosphorylation of Thr-1286 and Thr-1305 in the PEST motif can stabilize the ABCA1 protein.
In addition, a highly conserved motif (VFVNFA) located from positions 2215 to 2220 in the ABCA1 C-terminus is required for ABCA1 binding to helical apolipoproteins and mimic peptides. Alteration of this motif eliminates the binding of helical apolipoproteins and mimic peptides to ABCA1, and severely impairs ABCA1 protein stability induced by these apolipoproteins and peptides (Fitzgerald et al., 2004).
In brief, helical apolipoproteins and mimic peptides bind and stabilize the ABCA1 protein by several mechanisms. Due to the abundant existence of helical apolipoproteins in vivo, it is rationally speculated that helical apolipoproteins play a critical role in the stabilization of the ABCA1 protein under physiological conditions.
Fatty acids
Fatty acids are elevated in the situation of abnormal lipid metabolism, including insulin resistance, diabetes, and nonalcoholic steatohepatitis, which impairs the stability of the ABCA1 protein and alters its normal subcellular localization (Park et al., 2008; Kuang et al., 2009; Lee et al., 2011, 2012; Shao and Ford, 2012).
Unsaturated fatty acids such as palmitoleate, oleate, linoleate, and arachidonate must be activated to their CoA derivatives by the long-chain acyl-CoA synthetase (ACS) during their intracellular metabolism (Li et al., 2010). The inhibition or deficiency of ACS completely reverses unsaturated fatty acid-induced ABCA1 destabilization (Kanter et al., 2011, 2012). The conversion of unsaturated fatty acid CoA derivatives increases cellular AMP/ATP ratios and destabilizes the ABCA1 protein (Chen et al., 2005; Wang and Oram, 2007; Ellis et al., 2010). In addition, unsaturated fatty acids increase the phosphorylation of ABCA1 serine residues by activating a PLD2–PKCδ signaling pathway. Suppression of the serine phosphorylation completely abolishes ABCA1 destabilization (Wang and Oram, 2007; Yang et al., 2010; Ku et al., 2011). Thus, these findings suggest that unsaturated fatty acid-induced ABCA1 destabilization is dependent on the phosphorylation of the ABCA1 protein by the PKC pathway through the fatty acid CoA derivatives.
The saturated fatty acids, palmitate and stearate, can be desaturated by stearoyl-CoA desaturase (SCD), thereby destabilizing the ABCA1 protein. Fatty acids (two- to threefold higher, both saturated and unsaturated) and SCD are increased in normal macrophages exposed to diabetic sera. The SCD protein represents a potential link between the intracellular saturated fatty acids content and ABCA1 stability. Increased activity of SCD results in the conversion of saturated fatty acids to their monounsaturated equivalents and aggravates the destabilization of the ABCA1 protein. On the other hand, inhibition of SCD by conjugated linoleic acid and troglitazone essentially abolished palmitate and stearate-induced destabilization of ABCA1. These results suggest that SCD converts saturated fatty acids into ABCA1-destabilizing monounsaturated fatty acids, damaging the stability of the ABCA1 protein (Wang et al., 2004; Wong et al., 2011).
The destabilized ABCA1 protein by SCD is presumably composed of a negative feedback regulation in vivo. SCD may contribute to intracellular lipid accumulation by reducing the stability of the ABCA1 protein in macrophages. Recent studies demonstrated that diabetic dyslipidemia increases SCD expression and results in destabilization of the ABCA1 protein and reduction of ABCA1 levels in macrophages (Mauerer et al., 2009; Wong et al., 2011), which is predominantly due to raising unsaturated fatty acids. SCD deficiency in Ldlr-/- mice does show an anti-atherogenic profile, reducing plasma triglycerides and protecting from diet-induced obesity and insulin resistance. However, the mice show increased atherosclerosis due to increased inflammation (MacDonald et al., 2009).
PDZ proteins
Various PDZ proteins that are named for the founding members of the group (PSD-95, Dlg, ZO-1) and carry one or more copies of a 90-amino acid-binding domain are reported to interact with the consensus binding motif (S/T-X-V-COOH) present in the C-terminus of ABCA1 and increase ABCA1 stabilization. PDZ proteins such as β1-syntrophin, α1-syntrophin, and Lin7 interact with ABCA1 via the C-terminal-binding motif in ABCA1. Coexpression of syntrophins with ABCA1 stabilizes the ABCA1 protein and increases ABCA1 cell-surface expression (Munehira et al., 2004; Okuhira et al., 2005). The Rho guanine nucleotide exchange factors PDZ-RhoGEF and LARG also bind to the C terminus of ABCA1 and increase ABCA1 protein stability by activating RhoA (Okuhira et al., 2007; Fitzgerald et al., 2007; Okuhira et al., 2010). The underlying reason is probably that PDZ-RhoGTPases mediate the linkage of the ABCA1 protein to actin cytoskeleton, which then stabilizes the ABCA1 protein (Okuhira et al., 2005; Okuhira et al., 2010).
Other factors
Caveolin-1 enhances ABCA1 stability by interacting with ABCA1 in the plasma membrane and cytoplasm. Caveolin-1 suppression destabilizes the ABCA1 protein and decreases surface ABCA1 expression (Lin et al., 2007; Lin et al., 2009; Buechler and Bauer, 2011). Wogonin, one component in Scutellaria Baicalensis Georgi extracts, influences the phosphatase 2B (PP2B)-mediated ABCA1 dephosphorylation, and then increases ABCA1 stability and protein levels (Chen et al., 2010). The oxysterol-binding protein negatively regulates ABCA1 expression by decreasing ABCA1 protein stability in the cytoplasmic compartment (Bowden and Ridgway, 2008). Insulin destabilizes the ABCA1 protein by specifically inducing the phosphorylation of Tyr1206 in ABCA1 under the conditions of insulin resistance/hyperinsulinism (Tang et al., 2010; Nonomura et al., 2011).
The Degradation of ABCA1 Protein
The ABCA1 protein level is delicately manipulated by intracellular degradation mechanisms. An excessive and unqualified ABCA1 protein undergoes rapid proteolytic degradation. Intracellular degradation of the ABCA1 protein includes the calpain-mediated degradation pathway and the ubiquitin-mediated degradation pathway. (Fig. 2).
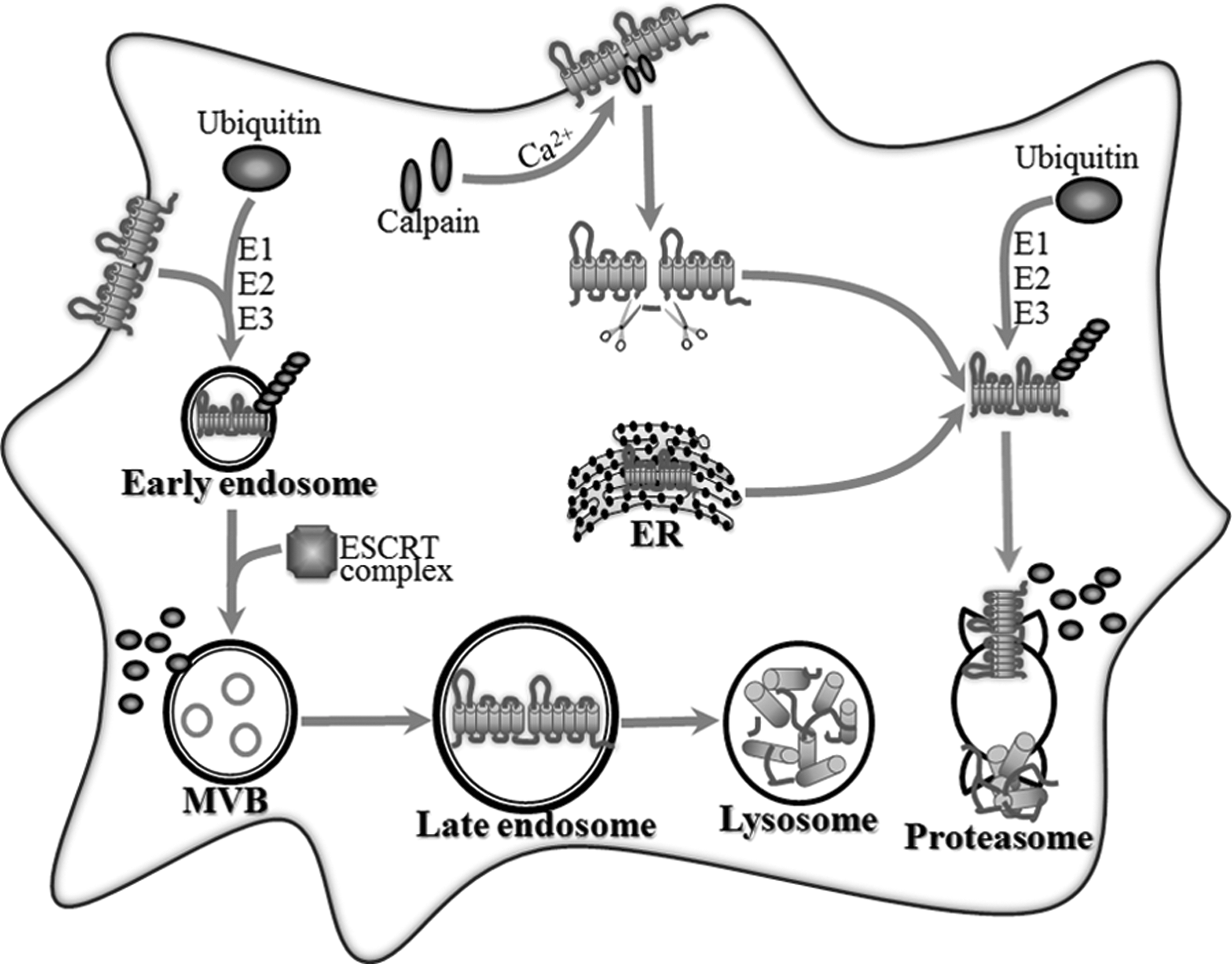
Schematic summary of the intracellular degradation pathway of the ABCA1 protein. The surface ABCA1 protein is mainly cleaved by calpain in a Ca2+-dependent manner. Two presumable positions are, respectively, in 1–5-8–14 and the PEST motif within the first intracellular loop. The ABCA1 remnant is internalized and completely degraded. The surface ABCA1 protein is also labeled by the ubiquitin chain with the catalysis of E1, E2, and E3. Then, ubiquitinated ABCA1 is sorted by the ESCRT complex and digested in lysosome through autophagy. Ubiquitin-mediated proteasomal degradation functions as the secondary quality control system to degrade intracellular aberrant ABCA1 originated from ER or ABCA1 remnant derived from calpain degradation. MVB, multivesicular body; E1, ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme; E3, ubiquitin-protein ligase; ESCRT, endosomal sorting complex required for transport.
The calpain-mediated degradation pathway is dependent on protease calpain to hydrolyze the ABCA1 protein (Miyazaki et al., 2013). A sequence located at amino acids 1283–1306 in the first intracellular loop of ABCA1 that is enriched in the PEST motif is required for the calpain-mediated ABCA1 degradation (Yokoyama et al., 2012). Two putative calpain cleavage positions are located in that domain. One is within the N-terminal domain of the 1–5-8–14 motif, and the other is within the PEST motif. The phosphorylation of Thr-1286 or Thr-1305 within the PEST motif destabilizes surface-resident ABCA1 and accelerates ABCA1 degradation by calpain protease, thereby reducing the cell surface localization and function of ABCA1 (Wang et al., 2003; Wang and Tall, 2003).
Ubiquitin mediates ABCA1 protein proteolysis via the lysosomal and nonlysosomal degradation pathway. Ubiquitin-mediated lysosomal degradation hydrolyzes cell surface-resident ABCA1 through the endosomal sorting complex required for the transport (ESCRT) pathway (Mizuno et al., 2011). The self-assembled ESCRT complex tethers ubiquitinated ABCA1 proteins and sorts them into vesicles, and then traffics these ubiquitinated cargoes to lysosomes for degradation (Raiborg and Stenmark, 2009; Boura et al., 2012). The disruption of the ESCRT pathway significantly delays the degradation of the cell surface ABCA1 protein. Ubiquitin-mediated nonlysosomal degradation is involved in ABCA1 protein clearance through the ubiquitin-proteasome system (Willis et al., 2010; Ogura et al., 2011). Suppressing proteasomes leads to an accumulation of ubiquitinated ABCA1 and an elevation of the total ABCA1 protein. Moreover, the ubiquitin-mediated nonlysosomal degradation pathway functions as a secondary quality control system to degrade ubiquitinated ABCA1 derived from the ER (Mizuno et al., 2011).
The degradation pathways mentioned above synergistically control the level of the ABCA1 protein residing on plasma membranes and in endosomes. So far, diverse factors have been proven to modulate ABCA1 protein levels by acting on ABCA1 degradation pathways. (Table 2)
Calmodulin
Calmodulin, an acidic Ca2+-binding protein, interacts with the ABCA1 protein directly and indirectly. Calmodulin directly binds to the 1–5–8–14 motif that is near the PEST motif and located between amino acid residues 1245 to 1257 within the cytoplasmic loop of ABCA1 and stabilizes ABCA1 in a Ca2+-dependent manner. The presence of calmodulin/Ca2+ protects ABCA1 peptides from the calpain-mediated ABCA1 degradation, resulting in an increase in the level and function of ABCA1 (Yokoyama et al., 2012). Calmodulin knockdown increases the degradation of ABCA1. However, W7 a common calmodulin antagonist that binds to calmodulin/Ca2+ and blocks the interaction between calmodulin/Ca2+ and its target proteins somehow increases the binding of calmodulin to ABCA1 and protects it from calpain-mediated degradation (Iwamoto et al., 2010). Calmodulin also indirectly stabilizes the ABCA1 protein. The Ca2+-dependent calmodulin/calcineurin/JAK2 pathway improves the apoA-I-binding activity of ABCA1, and then stabilizes the transporter (Karwatsky et al., 2010; Mulay et al., 2011; Yokoyama et al., 2012; Zhao et al., 2012). ApoA-I is proven to induce Ca2+ influx into cells. The rise in cytosolic Ca2+ triggers Ca2+ binding to calmodulin, which causes the activation of calcineurin, and in sequence, stimulates JAK2 autophosphorylation.
COP9 signalosome
COP9 signalosome (CSN) strictly controls ubiquitination and deubiquitination of the ABCA1 protein, and then regulates the lysosomal and nonlysosomal degradation of the ABCA1 protein. The CSN complex consists of eight subunits (CSN1-CSN8) (Kato and Yoneda-Kato, 2009). CSN2 enhances CSN assembly and stabilizes the CSN complex. CSN5 and CSN6 subunits are the active centers involved in the deubiquitination activity in the CSN complex. CSN2 overexpression decreases ubiquitinated forms of the ABCA1 protein (Azuma et al., 2009; Ogura et al., 2011). CSN5 has been shown to interact with the ABCA1 protein and promote deubiquitination of ABCA1. Dysfunction of the CSN accelerates ubiquitin-mediated degradation, resulting in abnormal ABCA1 turnover.
Other factors
Ginkgo biloba extract (EGb761) increases the ABCA1 protein by reducing the calpain activity (Tsai et al., 2010; Buechler and Bauer, 2011; Lee et al., 2011). Resistin downregulates the ABCA1 protein through enhancing proteasome-mediated protein degradation (Lee et al., 2009). The HIV-1 Nef protein binds to the ABCA1 C-terminal amino acids and accelerates ABCA1 protein degradation through the proteasomal degradation pathway (Mujawar and Fitzgerald, 2009; Mujawar et al., 2010; Feeney et al., 2012). In summary, these factors affect the levels of ABCA1 protein by regulating its degradation.
The Activity of ABCA1 Protein
Some molecules and functional domains do not modulate ABCA1 expression, but alter the activity of ABCA1 (Okuhira, 2012)(Table 2). ATP binding and hydrolysis in both nucleotide-binding domains (NBD) are associated with the increased ABCA1 activity. NBD mutations lead to ABCA1 activity loss (Nagao et al., 2012). Inhibition of the ATPase activity of ABCA1 by vanadate, beryllium fluoride, or the absence of magnesium ions efficiently impairs the ABCA1 activity (Tanaka et al., 2003; Takahashi et al., 2006). Glibenclamide also suppresses the ABCA1 ATPase activity and hinders ABCA1-mediated and apoA-I-dependent cholesterol efflux (Takahashi et al., 2006). It has been reported that the ATPase activity of purified ABCA1 reconstituted in liposomes is reduced by the addition of cholesterol. On the other hand, phospholipids with choline head groups, such as phosphatidylcholine and sphingomyelin, preferentially stimulate the ABCA1 ATPase activity and increase its lipid efflux activity (Takahashi et al., 2006). These molecules modulate the lipid efflux activity of ABCA1 through regulating the ATPase activity of ABCA1 without altering the mRNA and protein levels of the transporter.
Conclusions and Perspectives
As summarized in this review, the posttranscriptional regulations play a significant part in the modulation of ABCA1 expression and lipid efflux function. The regulatory pathways include the clearance of ABCA1 mRNA and the trafficking, stability, degradation, and activity of the ABCA1 protein. Undoubtedly, these posttranscriptional regulations coordinately regulate ABCA1 expression in vivo. We suppose that a reasonable balance between the facilitative and inhibitive posttranscriptional regulations tightly controls ABCA1 protein levels and intracellular cholesterol homeostasis under normal physiological conditions. However, inhibitive posttranscriptional regulations likely downregulate ABCA1 expression in pro-atherosclerotic circumstance. For instance, ABCA1 mRNA clearance is accelerated by microRNAs and ABCA1 protein degradation is exacerbated by calpain and ubiquitin. ABCA1 suppression impairs its cholesterol efflux function and facilitates cholesterol accumulation and atherogenesis. To prevent cholesterol accumulation and atherosclerosis, the stimulation of facilitative posttranscriptional regulations and the suppression of inhibitive posttranscriptional regulations become effective measures to heighten ABCA1 expression. Based on this, many exogenous and endogenous substances have been discovered to upregulate ABCA1 expression. These substances, including mimic peptides, PDZ proteins, and calmodulin, have been shown to potently improve ABCA1 expression and lipid efflux ability in preliminary studies. Since the effects of these substances have been analyzed in isolated cultured cells, endosomatic studies in animals and carefully designed clinical trials will be a future requirement to understand the influences and mechanisms of these substances on ABCA1 protein levels and on whole body lipid homeostasis. Increasing the ABCA1 protein through posttranscriptional pathways will promote cholesterol efflux, prevent foam cell formation, and suppress or even reverse the atherosclerosis progress. These fundamental mechanisms may provide novel strategies to posttranscriptionally elevate ABCA1 expression, which can enhance anti-atherogenic lipid efflux via ABCA1 and reduce cardiovascular disease.
Footnotes
Acknowledgments
The authors are grateful to MD Anying Chen for providing literal revision. This work was financially supported by the National Natural Sciences Foundation of China (81070220 and 81170278), Aid Program for Science and Technology Innovative Research Team in Higher Educational Institutions (2008-244) of Human Province, and A Project Supported by Scientific Research Fund of Hunan Provincial Education Department (12C0339), China.
Disclosure Statement
We have no actual or potential competing financial interests.